WO2009063504A2 - Novel crystal modification of epinastine or salts thereof and process for preparation thereof - Google Patents
Novel crystal modification of epinastine or salts thereof and process for preparation thereof Download PDFInfo
- Publication number
- WO2009063504A2 WO2009063504A2 PCT/IN2008/000612 IN2008000612W WO2009063504A2 WO 2009063504 A2 WO2009063504 A2 WO 2009063504A2 IN 2008000612 W IN2008000612 W IN 2008000612W WO 2009063504 A2 WO2009063504 A2 WO 2009063504A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- epinastine
- hcl
- solution
- group
- solvent selected
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
Definitions
- the present invention relates to novel crystal modification of 3-amino-9,13b-dihydro-lH- dibenz-[c,f
- the present invention further relates to polymorphs of Epinastine hydrochloride salt (IA) and process for preparation thereof.
- Epinastine (3-amino-9,13b-dihydro-lH-dibenz-[c,f]imidazo[l,5-a]-azepine) belongs to the 2-aminoimidazolines and is a therapeutically active substances characterized primarily by its anti-allergenic and anti-histaminergic activity therapeutically in EP35749. It is used as the hydrochloride salt represented by formula IA.
- US 4313931 discloses 3-amino-9,13b-dihydro-lH- dibenz-[c,fjimidazolo[l,5-a]-aze ⁇ ine (Epinastine), non-toxic pharmacologically acceptable acid addition salt thereof and process for preparing the same.
- This patent also discloses pharmaceutical composition containing Epinastine or its salts and method for treating bronchial asthma and allergic bronchitis.
- the '931 patent does not discloses or discuss about the solid-state characterization of Epinastine base or its HCl salt. XRPD pattern of the Epinastine HCl obtained by this process is disclosed in JP 2004-300042.
- US 5312916 relates to process for preparing 3-amino-9,13b-dihydro-lH-dibenz[c,fJimi- dazolo[l,5-a]-azepine hydrochloride using dimethylformamide with no disclosure about solid state properties of the isolated product of Epinastine hydrochloride.
- US 6403790 relates to a process for preparing Epinastine hydrochloride in the high-melting crystal modification using water as solvent.
- the 790 patent also discloses that Epinastine hydrochloride exists in low melting crystal modification which melts at 250-263 0 C and high melting crystal modification which melts at 275-281 0 C.
- JP 2004-300042 discloses a crystal polymorph of Epinastine HCl 5 which is characterized by XRPD.
- the patent also discloses the method for producing the crystal polymorph which involves crystallizing high quality epiastin hydrochloride from an aprotic polar solvent.
- XRPD pattern of the Epinastine HCl obtained by following the process disclosed in US 4313931 is also disclosed in this patent.
- polymorphism exhibits polymorphisms and can exist in many crystalline forms.
- Polymorphism is the ability of the compound to exhibit more than one orientation or conformation of molecule within the crystal lattice.
- Drug substance exists in various polymorphic forms; differ from each other in terms of their stability, solubility, compressibility, flowability and spectroscopic properties, thus affecting dissolution, bioavailability and handling characteristics of the substance.
- Epinastine hydrochloride All the polymorphic forms of Epinastine hydrochloride reported in the prior art are crystalline in nature. It is known that the amorphous form of drug substance exhibits different dissolution characteristic. Amorphous forms can have solubility, several hundred times than that of the crystalline counterparts. Thus the present invention also provides novel amorphous form of Epinastine and hydrochloride salt thereof.
- the object of the present invention is to provide novel crystalline forms, Form I and Form II of Epinastine base and process of preparation thereof.
- Another object of the invention is to provide novel amorphous form of Epinastine base and process for preparation thereof.
- Yet another object of the invention is to provide seven novel crystalline forms, Form II, Form III, Form IV, Form V, Form VI, Form VII and Form VIII of Epinastine HCl and process for preparing them.
- Another object of the present invention is to provide novel amorphous form of Epinastine HCl and process for preparation thereof.
- Still further object of the present invention is to provide pharmaceutical composition containing novel crystalline or amorphous forms of Epinastine base or its hydrochloride salt.
- Another object of the present invention provides method of treatment of anti-allergic and anti-histaminergic activity with therapeutically effecting amount of the pharmaceutical composition comprising novel crystalline or amorphous forms of Epinastine base or its hydrochloride salt.
- novel crystalline forms, Form I and Form II and amorphous form of Epinastine base and process of preparation thereof is disclosed.
- the present invention provides seven crystalline forms, Form II, Form III, Form IV, Form V, Form VI, Form VII and Form VIII and amorphous form of Epinastine HCl and process for preparation thereof.
- the present invention provides pharmaceutical composition comprising novel crystalline or amorphous forms of Epinastine base or its hydrochloride salt.
- the present invention provides method of treatment of antiallergic and anti-histaminergic activity with therapeutically effecting amount of the pharmaceutical composition comprising novel crystalline or amorphous forms of Epinastine base or its hydrochloride salt.
- FIG. 1 is a characteristic X-ray Powder diffraction pattern of Epinastine base Form I.
- FIG. 2 is a characteristic X-ray Powder diffraction pattern of Epinastine base Form II.
- FIG. 3 is characteristic X-ray Powder diffraction pattern of Amorphous Epinastine Base.
- FIG. 4 is a characteristic X-ray Powder diffraction pattern of Epinastine HCl Form II.
- FIG. 5 is a characteristic X-ray Powder diffraction pattern of Epinastine HCl Form III.
- FIG. 6 is a characteristic X-ray Powder diffraction pattern of Epinastine HCl Form IV.
- FIG. 1 is a characteristic X-ray Powder diffraction pattern of Epinastine base Form I.
- FIG. 2 is a characteristic X-ray Powder diffraction pattern of Epinastine base Form II.
- FIG. 3 is characteristic X-ray Powder diffraction pattern of A
- FIG. 7 is a characteristic X-ray Powder diffraction pattern of Epinastine HCl Form V.
- FIG. 8 is a characteristic X-ray Powder diffraction pattern of Epinastine HCl Form VI.
- FIG. 9 is a characteristic X-ray Powder diffraction pattern Epinastine HCl Form VII.
- FIG. 10 is a characteristic X-ray Powder diffraction pattern of Epinastine HCl Form VIII.
- FIG.11 is a characteristic X-ray Powder diffraction pattern of Amorphous Epinastine HCl.
- the present invention describes novel crystalline forms, Form I and Form II of Epinastine base and seven crystalline forms, Form II, Form III, Form IV, Form V, Form VI, Form VII and Form VIII of Epinastine HCl.
- process for preparation of crystalline forms of Epinastine base and its hydrochloride salt there is also provided process for preparation of crystalline forms of Epinastine base and its hydrochloride salt.
- the present invention further provides amorphous form of Epinastine base and its hydrochloride salt and process for preparation thereof.
- Form I Epinastine base characterized by the XRPD peaks given below:
- Another embodiment of the invention provides process for preparing the crystalline Epinastine base Form I comprising the steps of, a) dissolving Epinastine base in solubilizing solvent; b) cooling and stirring the obtained solution; c) isolating the separated product .
- the solubilizing solvent is selected from a group comprising aliphatic ketones, nitriles and C1-C4 alcohols.
- the ketone is selected from the group consisting of acetone, ethyl methyl ketone, diethyl ketone preferably acetone.
- Nitrile used is acetonitrile and Ci-C 4 alcohols are selected from group consisting of methanol, ethanol, 1- propanol, isopropyl alcohol (IPA), butanol preferably IPA.
- Epinastine base is dissolved at reflux temperature of the solvent selected for dissolution or at temperature of 60-80°C.
- the obtained solution is cooled followed by stirring at temperature 5-1O 0 C for 2-8 hours.
- the separated solid is then isolated by filtration followed by drying at temperature range of 30-90 0 C preferably 65 0 C to get Epinastine base Form I.
- process for preparation of crystalline Epinastine base Form I comprises the following steps; a) dissolving/suspending Epinastine base in a solubilizing solvent; b) adding anti-solvent to the obtained solution; c) isolating the separated solid.
- the solubilizing solvent is selected from group comprising polar aprotic solvent, aliphatic cyclic ethers, aliphatic ketone and chlorinated hydrocarbons or mixtures thereof.
- the polar aprotic solvent is selected from dimethyl formamide (DMF), dimethyl sulfoxide (DMSO), dimethyl acetamide (DMA).
- the aliphatic cyclic ether is selected from 1,4 - dioxane or tetrahydrofuran (THF) or mixtures thereof, preferably 1,4-dioxane.
- the aliphatic ketone is j acetone.
- the chlorinated hydrocarbon is selected from chloroform or methylene dichloride (MDC) preferably MDC.
- the anti-solvent is selected from the group comprising of aliphatic ketones is selected from acetone, 2-butanone, diethyl ketone or mixtures thereof, preferably acetone, esters is selected from methyl acetate, ethyl acetate, butyl acetate or mixtures thereof, preferably ethyl acetate.
- hydrocarbon is selected from n-hexane and n-heptane preferably n-hexane
- ethers is selected from diethyl ether (DEE), diisopropyl ether (DEPE), methyl tert-butyl ether (MTBE) preferably DIPE
- aromatic hydrocarbons is selected from toluene and xylene preferably toluene and water.
- Epinastine base is dissolved at reflux temperature of individual solvent selected for dissolution or at temperature 40-80 0 C.
- the anti-solvent is added to the obtained solution and then stirred at temperature range of 0- 3O 0 C preferably 25-3O 0 C for several hours.
- the separated solid is isolated by filtration followed by drying at temperature 65 0 C to get Epinastine base Form I.
- Another embodiment of the present invention provides crystalline Epinastine base Form II characterized by the following XRPD peaks which are shown in the table below:
- process for preparation of Epinastine base Form II comprises the following steps, a) dissolving Epinastine base in a solubilizing solvent to obtain a solution; b) pouring the solution obtained in anti-solvent; c) stirring the solution for several hours and d) isolating the product .
- the solubilizing solvent is selected from group comprising alcohols, aliphatic cyclic ethers or mixtures thereof.
- the alcohol is selected from methanol, ethanol, 1-propanol, 2- propanol (IPA), butanol more preferably IPA.
- Aliphatic cyclic ether is selected from 1,4- dioxane, tetrahydrofuran preferably 1,4-dioxane.
- the anti-solvent selected is any suitable solvent which is miscible with the above solubilizing solvents most preferably water.
- the anti-solvent is cooled to -10 to 35 0 C preferably 0-5°C.
- Epinastine base is dissolved at reflux temperature of individual solvent selected for dissolution or at temperature 60-80°C.
- the obtained solution is then poured into anti-solvent at temperature of 0-10 0 C and stirred at the same temperature for 2-4 hours.
- the separated solid is isolated by filtration followed by drying at 65 0 C to get Epinastine Form II.
- the present invention provides novel amorphous form of Epinastine.
- the amorphous Epinastine is characterized by, having broad x-ray diffraction spectrum as in figure 3.
- Another embodiment of the present invention provides process for the preparation of amorphous Epinastine which comprises dissolving Epinastine base in a suitable solubilizing solvent at reflux temperature of suitable solvent selected for dissolution or at temperature range of 20-50 0 C and the solvent is removed from the clear solution by vacuum drying or spray drying technique to get amorphous Epinastine .
- the solubilizing solvent is selected from alcohol, chlorinated hydrocarbons or mixture thereof.
- the alcohol is selected from methanol, ethanol, preferably methanol.
- the chlorinated hydrocarbon is selected from methylene dichloride (MDC) or chloroform preferably methylene dichloride.
- the concentration of Epinatin base is 5-15% preferably 8-12 % w/v.
- the spray drying is carried out at inlet temperature of 40 to 170 0 C, preferably 160 0 C and outlet temperature of 35-85°C, preferably 65 0 C.
- the vacuum evaporation is carried out at temperature about 65-85°C.
- Epinastine HCl obtained by following the process disclosed in US 4313931 is designated herein as 'Form F of Epinastine hydrochloride.
- the present invention provides new form of Epinastine hydrochloride designated as Form II characterized by the XRPD peaks which are shown in the table below:
- the process for preparing Epinastine hydrochloride Form II comprises the steps of, a) suspending Epinastine base in solubilizing solvent b) adding acid to the obtained solution; c) warming the solution to get solution and d) isolating the separated solid.
- Epinastine base can be of any polymorphic form used for making novel form of Epinastine HCl.
- the solubilizing solvent is aqueous solvent selected such that Epinastine base is insoluble but its hydrochloride salt is freely soluble, preferably water.
- the acid used is hydrochloric acid, preferably used in concentrated form, the term "concentrated form” meaning an approximately 32% by weight aqueous hydrochloric acid.
- Epinastine base is suspended in solubilizing solvent and acid is added to the obtained suspension .
- the solution is warmed at reflux temperature or at about 50-60°C to get clear solution.
- the clear solution is then filtered and cooled to -10 to 5 0 C preferably 0-5°C.
- the suspension is further stirred at temperature 10-15 0 C for 30 min.
- the separated solid is then filtered and dried at 25-3O 0 C to get Epinastine HCl Form II.
- the present invention provides new form of Epinastine hydrochloride designated as Form III characterized by the following XRPD peaks which are shown in the table below:
- Another embodiment of the present invention provides process for preparing Epinastine hydrochloride Form III comprising the steps of, a) dissolving Epinastine hydrochloride in a solubilizing solvent; b) adding suitable antisoivmt; c) isolating the product and drying
- Another embodiment of the present invention provides process for preparing Epinastine hydrochloride Form III comprising the steps of, a)dissolving Epinastine hydrochloride in a solubilizing solvent; b)isolating the product and drying.
- Epinastine hydrochloride selected for making novel form of Epinastine hydrochloride can be of any polymorphic forms preferably Form II.
- the solubilizing solvent is selected from the group comprising of C 1 -Gt alcohol, chlorinated hydrocarbon and aliphatic cyclic ether.
- the C1-C4 alcohol is selected from the group of methanol, ethanol, 1-propanol, isopropyl alcohol, butanol preferably 2-propanol.
- Chlorinated hydrocarbons selected are methylene dichloride (MDC) or chloroform preferably MDC.
- the aliphatic cyclic ether is selected from 1,4-dioxane or tetrahydrofuran (THF).
- the anti-solvent is selected from group of aliphatic acyclic ether, hydrocarbon and ester.
- the aliphatic acyclic ether is selected from DEE, DIPE or MTBE preferably DIPE.
- Hydrocarbon is selected from pentane, hexane, heptane preferably hexane and ester is selected from ethyl acetate, butyl acetate, isopropyl acetate preferably ethyl acetate.
- the anti-solvent is added at 3O-5O°C preferably 25-35°C.
- the suspension is further cooled and stirred at 5-1O 0 C for 2-4 hours.
- the separated solid is filtered and dried at 90°C to get the novel Form III of Epinastine hydrochloride.
- Epinastine hydrochloride is dissolved in suitable solubilizing solvent at reflux temperature or at temperature 50-60 0 C.
- the anti- solvent is added at 30-50 0 C preferably 25-35 0 C.
- the suspension/solution is further stirred at 5-10 0 C for 2-4 hours.
- the separated solid is filtered and dried at 9O 0 C to get the novel Form III of Epinastine hydrochloride.
- the process for preparing Epinastine hydrochloride Form IV comprises the steps of, a) dissolving Epinastine HCl in a solubilizing solvent; b) adding antisolvent to the obtained solution; c) isolating the product and drying.
- Epinastine hydrochloride selected for making novel form of Epinastine hydrochloride can be of any polymorphic form, preferably Form II.
- the solubilizing solvent is selected ftom the group of C1-C4 alcohol or 1,4-cyclohexane.
- the alcohol is selected are methanol, ethanol, 2-propanol, preferably methanol.
- the antisolvent is selected from group of aliphatic acyclic ether, aromatic hydrocarbon and ester or mixtures thereof.
- the aliphatic acyclic ether is selected from DEE, DIPE or MTBE preferably DIPE.
- Aromatic hydrocarbon is selected from toluene, xylene preferably toluene and ester is selected from ethyl acetate, butyl acetate, iso propyl acetate preferably ethyl acetate.
- the anti-solvent is added at 30-60 0 C preferably 25-35°C.
- the suspension is further stirred at 5-10°C for 2-4 hours.
- the separated solid is filtered and dried at 50-65°C to get the novel form, Form IV of Epinastine hydrochloride.
- Epinastine hydrochloride is dissolved in suitable solubilizing solvent at reflux temperature or at temperature 50-60°C.
- the anti- solvent is added at 30-50 0 C preferably 25-35 0 C.
- the suspension is further stirred at 5-1O 0 C for 2-4 hours.
- the separated solid is filtered and dried at 50-65 0 C to get the novel Epinastine hydrochloride Form IV .
- the present invention provides new form of Epinastine hydrochloride, designated as Form V characterized by the following XRPD peaks which are shown in the table below:
- the process for preparing Epinastine hydrochloride Form V comprises the steps of, a) dissolving Epinastine HCl in solubilizing solvent; b) adding anti-solvent to the obtained solution; c) stirring the suspension for several hours; d) isolating the product and drying.
- the present invention provides process for preparation of Epinastine hydrochloride Form V which comprises suspending Epinastine hyrochloride (in any form) in suitable solubilizing solvent and isolating the product.
- Epinastine hydrochloride selected for making novel form of Epinastine hydrochloride can be of any polymorphic forms preferably Form II.
- the solubilizing solvent is selected from C1-C4 alcohol such as methanol, ethanol, 2- propanol, preferably ethanol and aromatic hydrocarbon such as toluene, xylene preferably toluene.
- the anti-solvent is selected from a group of aliphatic acyclic ether such as DEE, DIPE or MTBE.
- the anti-solvent is cooled to -10 to 35°C preferably 10-25°C.
- Epinastine hydrochloride is dissolved in suitable solubilizing solvent at reflux temperature or at temperature of 50-60 0 C.
- the anti- solvent is added at 30-50°C preferably 25-35°C.
- the suspension is further cooled and stirred at 5-l0°C for 2-4 hours.
- the separated solid is filtered and dried at 50-65 0 C to get the novel form, Epinastine hydrochloride Form V.
- the present invention provides new form of Epinastine hydrochloride designated as Form VI characterized by the following XRPD peaks which are shown in the following table below,
- the process for preparing Epinastine hydrochloride Form VI comprises the steps of, a) dissolving Epinastine HCl in solubilizing solvent; b) adding antisolvent to the obtained solution; c) isolating the product and drying.
- Epinastine hydrochloride selected for making novel form of Epinastine hydrochloride can be of any polymorphic forms preferably Form II.
- the solubilizing solvent is selected from the group of C1-C4 alcohols, cyclic ethers and chlorinated hydrocarbons.
- the alcohol selected are methanol, ethanol, 2-propanol, preferably ethanol.
- the aliphatic cyclic ether selected are 1,4-dioxane, THF preferably 1,4-dio ⁇ ane and chlorinated hydrocarbon selected are MDC, chloroform preferably MDC.
- the anti-solvent is selected from a group of aliphatic acyclic ethers or hydrocarbons.
- the aliphatic acyclic ether is selected from DEE 5 DIPE or MTBE and aliphatic hydrocarbon is selected from pentane, hexane or heptane preferably n-hexane.
- Epinastine hydrochloride is dissolved in suitable solubilizing solvent at reflux temperature or at temperature 50-60°C.
- the anti- solvent is added at 30-50°C preferably 25-35 0 C.
- the suspension is further stirred at 5-1O 0 C for 2-4 hours.
- the separated solid is filtered and dried at 50-65 0 C to get the novel Form VI of Epinastine hydrochloride.
- the present invention provides new form of Epinastine hydrochloride designated as Form VII characterized by the following XRPD peaks which are shown in the table below, Pos. r°2Th.l ReI. Int. ⁇ % ⁇
- the process for preparing Epinastine hydrochloride Form VII comprises the steps of, a) dissolving Epinastine HCl in a solubilizing solvent; b) adding anti-solvent to the obtained solution; c) stirring the solution for several hours; d) isolating the product and drying
- Epinastine HCl selected for making novel form of Epinastine HCl can be of any polymorphic forms preferably Form II.
- the solubilizing solvent is selected from the group of cyclic ether and polar aprotic solvent.
- the aliphatic cyclic ether is selected from 1,4-dioxane, tetrahydrofuran (THF) preferably 1,4-dioxane and polar aprotic solvent is selected from dimethyl formamide (DMF), dimethyl sulfoxide (DMSO), N,N-dimethyI acetamide (DMA) preferably DMF.
- the anti-solvent is selected from a group of aliphatic ketones and esters.
- the aliphatic ketone is selected from acetone, 2-butanone, diethyl ketone preferably acetone and ester is selected from ethyl acetate, butyl acetate, iso propyl acetate, preferably ethyl acetate.
- Epinastine hydrochloride is dissolved in suitable solubilizing solvent at reflux temperature or at temperature 50-60°C.
- the anti- solvent is added at 30-50 0 C preferably 25-35 0 C.
- the suspension is further stirred at 5-10 0 C for 2-4 hours.
- the separated solid is filtered and dried at 50-65 0 C to get the novel Form VII of Epinastine hydrochloride.
- the present invention provides new form of Epinastine hydrochloride designated as Form VIII characterized by the following XRPD peaks which are shown in the table below,
- the process for preparing Epinastine hydrochloride Form VIII comprises the steps of, a) dissolving Epinastine HCl in a solubilizing solvent b) adding antisolvent to the obtained solution; c) stirring the suspension for several hours. d) isolating the product and drying
- the process for preparation of Epinsatine HCl Form VIII comprises dissolving Epinastine HCl in a solubilizing solvent and stirring the obtained solution at 5-1O 0 C for several hours preferably 2-6 hours to get the solid of Epinsatine HCl Form VIII.
- the process for preparing Epinastine hydrochloride Form VIII comprises the steps of, e) dissolving Epinastine HCl in a solubilizing solvent f) pouring the obtained solution to anti-solvent ; g) stirring the suspension for several hours, h) isolating the product and drying
- Epinastine HCl selected for making novel form of Epinastine HCl can be of any polymorphic forms preferably Form II.
- the solubilizing solvent is selected from the group of chlorinated hydrocarbon, aliphatic cyclic ethers and nitrile.
- the chlorinated hydrocarbon is selected from methylenedichloride (MDC), chloroform preferably MDC, aliphatic cyclic ethers selected are 1,4-dioxane, THF preferably 1,4-dioxane and nitrile used is acetonitrile.
- the anti-solvent is selected from group of aliphatic ketone, esters and ether.
- the aliphatic ketone is selected from acetone, 2-butanone, diethyl ketone preferably acetone, ester selected is ethyl acetate, butyl acetate preferably ethyl acetate and ether is selected from DEE, DIPE and MTBE.
- the anti-solvent is cooled to -10 to 30 0 C preferably 20-25°C.
- Epinastine hydrochloride is dissolved in suitable solubilizing solvent at reflux temperature or at temperature 50-60°C to obtain the solution.
- the obtained solution is added to the cooled antisolvent or anti-solvent is added to the obtained solution at 30-50°C preferably 25-35 0 C.
- the suspension is further stirred at 5-10 0 C for 2-4 hours.
- the separated solid is filtered and dried at 50-65 0 C to get the novel Form VIII of Epinastine hydrochloride.
- the present invention provides a novel amorphous form of Epinastine HCl.
- the amorphous Epinastine hydrochloride in accordance with the present invention is characterized by having broad x-ray diffraction spectrum as in figure 11.
- Another embodiment of the present invention provides process for the preparation of amorphous Epinastine HCl which comprises dissolving Epinastine HCl in a suitable solubilizing solvent at a temperature range of 20-40 0 C and the solvent is removed from the obtained solution by vacuum drying or spray drying or lyophilization technique to get amorphous Epinastine HCl .
- the solubilizing solvent is selected from water, alcohol or mixtures thereof preferably water.
- the alcohol is selected from methanol and ethanol preferably methanol.
- the concentration of Epinastine HCl in solution is 5-15% preferably 8-12 %.
- the spray dried is carried out at inlet temperature of 40 to 170°C, preferably 160°C and outlet temperature of 35-85 0 C, preferably 65 0 C.
- the vacuum evaporation is carried out at temperature ranges from 65-85°C
- the lyophilization is carried out by freeze drying an aqueous solution at temperature -20 to -8O 0 C under vacuum preferably -40 0 C.
- room temperature refers to a temperature at about 25° C.
- the crystallization process hitherto described to prepare the novel polymorphs consists of dissolving Epinastine base or Epinastine HCl in the selected solvent either with or without heating, preferably with heating at or near boiling point of the solvent. Cooling the resultant solution to -10 0 C to 5°C for several hours to regenerate the solid. Isolating the precipitated solids and drying the isolated solids at about ambient to 65 0 C temperature.
- the solvent and anti-solvent combination process described to prepare the novel polymorphs consist of dissolving Epinastine base or Epinastine HCl in the suitable solvent.
- the dissolution may be carried out at room temperature or under reflux condition. Adding anti-solvent to the resulting solution under warm condition to get polymorphs of Epinastine base or Epinastine HCl.
- the anti-solvent addition is carried out at room temperature or at temperature of 25-35 0 C. Isolating the precipitated solids by filtration and drying the isolated solids at about ambient temperature to 65°C or at room temperature.
- a solution of aluminum hydride in tetrahydrofuran was produced by slowly adding drop wise solution of 275 gm of 98% sulfuric acid in 0,6 liters of anhydrous tetrahydrofuran to a suspension of 216 gm of lithium aluminum hydride in 7.5 liters of absolute tetrahydrofuran under stirring. Without separating Lithium sulphate that forms, a solution of 300 gm of 6-Cyano azepine in 2.4 liters tetrahydrofuran was added within 30 min. The reaction mixture was stirred for 2 hours at room temperature and then decomposed while maintaining temperature below 0 0 C of the hydride by addition of 0.75 liters of water. The inorganic salts were removed by suction filtration. The filtrate was evaporated under reduced pressure at 50-55°C completely to get dark reddish brown colored oil.
- the above prepared amino compound was dissolved in 1.5 L methanol at temperature 25-30 0 C .
- 112 gm of fumaric acid was added lot wise in 30-40 minutes at 35-40 0 C and the reaction mixture was stirred for 1 hour.
- the solvent was removed completely under vacuum at temperature below 60 0 C .
- 1.5 L acetonitrile was added to the obtained reaction mass and stirred for 25 to 30 minutes at temperature 25-30 0 C .
- the reaction mixture was filtered and suck dried.
- the wet cake of fumarate salt was suspended in 1.12 L water and sodium hydroxide solution (87 gm NaOH dissolved in 450 ml water) was added slowly at temperature 25-30 0 C followed by stirring for 30 minutes.
- Epinastine amine was extracted with 2x2.25 L MDC.
- the MDC layer was washed with water, dried on sodium sulphate and concentrated to get pure Epinastine amine. Yield: 200-205 gm (64-65% of Theory).
- Example C Preparation of 3-amino-9,13b-dihydro-lH-dibenzo[c,f]iinidazo[l,5-a] azepine hydrobromide: 365gm of 6-aminomethyl-6,ll-dihydro-5H-dibenzo[b,e]azepine was dissolved in 3.65 liters ethanol and a solution of 176 gm cyanogen bromide in 1.5 liters of absolute tetrahydrofuran was added drop wise under stirring at room temperature. The reaction mixture was stirred overnight at room temperature and then admixed with 2.74 liters of diethyl ether. Subsequently, resulted crystals were filtered and washed with 0.70 liters of diethyl ether.
- Example F Preparation of 3-amino-9,13b-dihydro-lH-dibenzo[c,f]imidazo[l,5-a] azepine HCl Form III: 100 gm of 3-ammo-9,13b-dihydro-lH-dibenzo[c,fJimidazo[l,5- a] azepine was suspended in 0.3 liters 1,4-dioxane and to this 41.8 ml cone. HCl was added. The clear solution obtained was rapidly added to 6.1 liters ethyl acetate followed by stirring for 30 minutes, filtered the solution at ambient temperature to isolate Epinastine HCl Form III.
- Example G Conversion of Form III of 3-amino-9,13b-dihydro-lH- dibenzo[c,fjimidazo[l,5-a] azepine HCI to Form VIII: 100 gm of 3-amino-9,13b- dihydro-lH-dibenzo[c,f]iinidazo[l,5-a] azepine HCl Form III was suspended in 1 liters acetonitrile and heated to reflux for 24 hrs under stirring to ensure complete transformation of Form III to Form VIII. The solution obtained was filtered at ambient temperature to isolate Epinastine HCl Form VIII.
- Epinastine HCl Form VIII obtained was suspended in 1 liter ethyl acetate and heated to reflux for 24 hrs under stirring to ensure concentration of acetonitrile below 400 ppm. The solution was filtered at ambient temperature to isolate Epinastine HCl Form VIII.
- Epinastine base 0.5g was dissolved in 10 ml isopropyl alcohol at reflux. The solution was filtered to remove any insoluble material. The solution was cooled and stirred at 5-1O 0 C for 3 hrs. The separated solid was isolated by filtration and dried at 65°C to get Epinastine Form I.
- Epinastine base 0.5g was dissolved in 60 ml acetonitrile at reflux. The solution was filtered to remove any insoluble material. The solution was cooled and stirred at 5-10°C for 3 hrs. The separated solid was isolated by filtration and dried at 65 °C to get Epinastine Form I.
- Epinastine base 0.5g was dissolved in 6 ml dimethylsulphoxide (DMSO) at 55-60°C. The solution was filtered to remove any insoluble material. To this solution 30 ml acetone was added dropwise. The solution was stirred at 25-30°C for 3 hrs. The separated solid was isolated by filtration and dried at 65°C to get Epinastine Form I.
- DMSO dimethylsulphoxide
- Epinastine base 0.5g was dissolved in 6 ml DMSO at 55-6O 0 C. The solution was filtered to remove any insoluble material. To this solution 20 ml water was added dropwise. The solution was stirred at 25-30 0 C for 3 Hrs. The separated solid was isolated by filtration and dried at 65°C to get Epinastine Form I.
- Epinastine base 0.5g was dissolved in 10 ml methylenedichloride (MDC) at 40-45 0 C. The solution was filtered to remove any insoluble material. To this solution 30 ml DIPE was added dropwise. The solution was stirred at 25-30 0 C for 3 Hrs. The separated solid was isolated by filtration and dried at 65 0 C to get Epinastine Form I.
- MDC methylenedichloride
- Epinastine base 0.5g was dissolved in 10 ml MDC at 40-45 0 C. The solution was filtered to remove any insoluble material. To this solution 30 ml hexane was added dropwise. The solution was stirred at 25-3O 0 C for 3 Hrs. The separated solid was isolated by filtration and dried at 65 0 C to get Epinastine Form I.
- Epinastine base 0.5g was dissolved in 10 ml MDC at 40-45 0 C. The solution was filtered to remove any insoluble material. To this solution 50 ml Toluene was added dropwise. The solution was stirred at 25-3O 0 C for 3 Hrs. The separated solid was isolated by filtration and dried at 65°C to get Epinastine Form I.
- Epinastine base 0.5g was dissolved in 10 ml MDC at 40-45 0 C. The solution was filtered to remove any insoluble material. To this solution 30 ml ethyl acetate was added dropwise. The solution was stirred at 25-30 0 C for 3 Hrs. The separated solid was isolated by filtration and dried at 65°C to get Epinastine Form I.
- Epinastine 0.5g was base dissolved in 6 ml 1,4-dioxane at reflux. The solution was filtered to remove any insoluble material. To this solution 30 ml toluene was added dropwise. The solution was stirred at 5-10°C for 3 Hrs. The separated solid was isolated by filtration and dried at 65 0 C to get Epinastine Form I.
- Epinastine base 0.5g was dissolved in 6 ml 1,4-dioxane at reflux. The solution was filtered to remove any insoluble material. To this solution 40 ml acetone was added dropwise. The solution was stirred at 25-3O 0 C for 3 Hrs. The separated solid was isolated by filtration and dried at 65°C to get Epinastine Form I.
- Epinastine base 0.5g was dissolved in 6 ml 1,4-dioxane at reflux. The solution was filtered to remove any insoluble material. To this solution 25 ml hexane was added dropwise. The solution was stirred at 25-3O 0 C for 3 Hrs. The separated solid was isolated by filtration and dried at 65°C to get Epinastine Form I.
- Epinastine base 0.5g was dissolved in 6 ml 1,4-dioxane at reflux. The solution was filtered to remove any insoluble material. To this solution 35 ml ethyl acetate was added dropwise. The solution was stirred at 0-5 0 C for 3 Hrs. The separated solid was isolated by filtration and dried at 65°C to get Epinastine Form I.
- Epinastine base 0.5g was dissolved in 12 ml THF at reflux. The solution was filtered to remove any insoluble material. To this solution 30 ml hexane was added dropwise. The solution was stirred at 25-3O 0 C for 3 Hrs. The separated solid was isolated by filtration and dried at 65°C to get Epinastine Form I.
- Epinastine base 0.5g was dissolved in 12 ml THF at reflux. The solution was filtered to remove any insoluble material. To this solution 30 ml DIPE was added dropwise. The solution was stirred at 25-30 0 C for 3 Hrs. The separated solid was isolated by filtration and dried at 65°C to get Epinastine Form I.
- Epinastine base 0.5g was dissolved in 10 ml isopropylalcohol at reflux. The solution was filtered to remove any insoluble material. The clear solution was poured in 30 ml water at 0-5 0 C. The solution was stirred at 0-5 0 C for 3 Hrs. The separated solid was isolated by filtration and dried at 65°C to get Epinastine Form II.
- Epinastine base 0.5g was dissolved in 6 ml 1,4-dioxane at reflux. The solution was filtered to remove any insoluble material. The clear solution was poured in 30 ml water at 0-5 0 C. The solution was stirred at 0-5 0 C for 3 Hrs. The separated solid was isolated by filtration and dried at 65 0 C to get Epinastine Form II.
- Example 19 5 g Epinastine base was dissolved in 10 ml methanol at a temperature range of 40-50 0 C. The clear solution obtained was concentrated under vacuum at 65 0 C to get amorphous Epinastine.
- Example 20 5 g Epinastine base was dissolved in 10 ml methanol at a temperature range of 40-50 0 C. The clear solution obtained was concentrated under vacuum at 65 0 C to get amorphous Epinastine.
- Example 20 5 g Epinastine base was dissolved in 10 ml methanol at a temperature range of 40-50 0 C. The clear solution obtained was concentrated under vacuum at 65 0 C to get amorphous Epinastine.
- Epinastine base was dissolved in methanol at a temperature range of 30-40 0 C. Concentration of Epinastine used for spray drying was about 10 % weight/volume. Spray drying is carried out at the inlet temperature 120° C and outlet temperature 65 0 C to get amorphous Epinastine.
- Epinastine base was dissolved in MDC at a temperature range of 30-40 0 C. Concentration of Epinastine used for spray drying was about 10 % weight/volume. Spray drying is carried out at the inlet temperature 120° C and outlet temperature 65°C to get amorphous
- Epinastine HCl 0.5g was dissolved in 10 ml IPA at reflux temperature. To this clear solution 50 ml hexane was added and the solution was stirred at 5-10°C for 3 hrs. The separated solid was isolated by filtration and dried at 65 0 C to get Epinastine Form III.
- Epinastine HCl 0.5g was dissolved in 10 ml MDC at reflux temperature. To this clear solution 30 ml DIPE was added and the solution was stirred at 5-10°C for 3 hrs. The separated solid was isolated by filtration and dried at 65°C to get Epinastine Form III.
- Epinastine HCl 0.5g was dissolved in 15 ml MDC at reflux temperature. To this clear solution 30 ml ethyl acetate was added and the solution was stirred at 5-10°C for 3 Hrs. Separated solid was isolated by filtration and dried at 65°C to get Epinastine Form III.
- Epinastine HCI 0.5g was dissolved in 15 ml MDC at reflux temperature. The obtained solution was cooled and stirred at 5-10 0 C for 3 hrs. The separated solid was isolated by filtration and dried at 65°C to get Epinastine Form III.
- Epinastine HCl 0.5g was dissolved in 6 ml 1,4-dioxane at 50-60°. To this clear solution 20 ml ethyl acetate was added and the solution was stirred at 5-10 0 C for 3 hrs. The separated solid was isolated by filtration and dried at 65 0 C to get Epinastine Form III.
- Epinastine HCl 0.5g was suspended in 30 ml THF at reflux temperature. The solution was cooled and stirred at 5-10 0 C for 3 hrs. The separated solid was isolated by filtration and dried at 65°C to get Epinastine Form III.
- Example 31 0.5g Epinastine HCl was dissolved in 5 ml methanol at 50-60°. To this clear solution 30 ml DIPE was added and the solution was stirred at 5-10°C for 3 hrs. The separated solid was isolated by filtration and dried at 65°C to get Epinastine Form IV.
- Epinastine HCl was dissolved in 5 ml methanol at 50-60°. To this clear solution mixture of 40 ml DIPE and 40 ml toluene was added and the solution was stirred at 5-10°C for 3 hrs. The separated solid was isolated by filtration and dried at 65 0 C to get Epinastine Form IV.
- Epinastine HCl 0.5g was dissolved in 10 ml ethanol at 50-60°C. To this clear solution 40 ml DIPE was added and the solution was stirred at 5-10°C for 3 hrs. The separated solid was isolated by filtration and dried at 65°C to get Epinastine Form V.
- Epinastine HCl 0.5g was suspended in 30 ml toluene at reflux temperature. The solution was cooled and stirred at 5-10°C for 3 hrs. The separated solid was isolated by filtration and dried at 65°C to get Epinastine Form V.
- Epinastine HCl 0.5g was dissolved in 10 ml ethanol at reflux. To this clear solution 60 ml hexane was added and the solution was stirred at 5-10°C for 3 hrs and the separated solid was isolated by filtration and dried at 65 0 C to get Epinastine Form VI.
- Epinastine HCl 0.5g was dissolved in 15 ml MDC at reflux. To this clear solution 60 ml hexane was added and the solution was stirred at 5-10 0 C for 3 hrs. The separated solid was isolated by filtration and dried at 65 0 C to get Epinastine Form VI.
- Example 37 0.5g Epinastine HCl was dissolved in 6 ml 1,4-dioxane at 50-60°. To this clear solution add 30 ml DIPE. The solution was stirred at 5-10°C for 3 Hrs. Separated solid was isolated by filtration and dried at 65 0 C to get Epinastine Form VI.
- Epinastine HCl 0.5g was dissolved in 6 ml 1,4-dioxane at 50-60°. To this clear solution add 30 ml Acetone. The solution was stirred at 5-10°C for 3 Hrs. Separated solid was isolated by filtration and dried at 65 0 C to get Epinastine Form VII.
- Epinastine HCl was dissolved in 4 ml dimethylformamide (DMF) at 50-60°. To this clear solution add 20 ml Ethyl acetate. The solution was stirred at 5-1O 0 C for 3 Hrs. Separated solid was isolated by filtration and dried at 65 0 C to get Epinastine Form VII.
- DMF dimethylformamide
- Epinastine HCl 0.5g was dissolved in 15 ml MDC at reflux. To this clear solution add 30 ml Acetone. The solution was stirred at 5-10°C for 3 Hrs. Separated solid was isolated by filtration and dried at 65°C to get Epinastine Form VIII.
- Epinastine HCl 0.5g was dissolved in 10 ml acetonitrile at reflux! From the clear solution solid separates out at reflux. The solution was stirred at 5-10°C for 3 Hrs. Separated solid was isolated by filtration and dried at 65°C to get Epinastine Form VIII.
- Epinastine HCl was dissolved in 10 ml 1,4-dioxane at 50-60°. The clear solution obtained is added to 40 ml Ethyl acetate. The solution was stirred at 5-1O 0 C for 3 Hrs. Separated solid was isolated by filtration and dried ait 65 0 C to get Epinastine Form VIII.
- Epinastine HCl 5 g was dissolved in 10 ml water at a temperature range of 40-50 0 C. The clear solution is concentrated under vacuum at 85°C to get Amorphous Epinastine HCl.
- Epinastine HCl was dissolved in 20 ml methanol at a temperature range of 40-50 0 C.
- Epinastine HCl was dissolved in water at a temperature range of 30-40 0 C. Concentration of Epinastine used for spray drying is about 10 % weight/volume. Spray drying was carried out at the inlet temperature 140° C and outlet temperature 65 0 C to get Amorphous Epinastine HCl.
- Epinastine HCl was dissolved in methanol at a temperature range of 30-40 0 C. Concentration of Epinastine used for spray drying is about 10 % weight/volume. Spray drying was carried out at the inlet temperature 120° C and outlet temperature 65°C to get Amorphous Epinastine HCl.
- Epinastine HCl was dissolved in 100 ml water at a temperature range of 40-50 0 C. The clear solution was subjected to lyophilization for 24-48 hrs. to get Amorphous Epinastine HCl.
Abstract
The present invention provides novel crystalline forms of 3-amino-9,13b-dihydro-lH- dibenzo[c,f]imidazo[l55-a] azepine or salts thereof and process for preparation thereof.
Description
Technical field:
The present invention relates to novel crystal modification of 3-amino-9,13b-dihydro-lH- dibenz-[c,f|imidazo[l,5-a]-azepine (Epinastine base) of formula (I) and process for preparation thereof. The present invention further relates to polymorphs of Epinastine hydrochloride salt (IA) and process for preparation thereof.

Formula (I) Background of the invention:
Epinastine (3-amino-9,13b-dihydro-lH-dibenz-[c,f]imidazo[l,5-a]-azepine) belongs to the 2-aminoimidazolines and is a therapeutically active substances characterized primarily by its anti-allergenic and anti-histaminergic activity therapeutically in EP35749. It is used as the hydrochloride salt represented by formula IA.
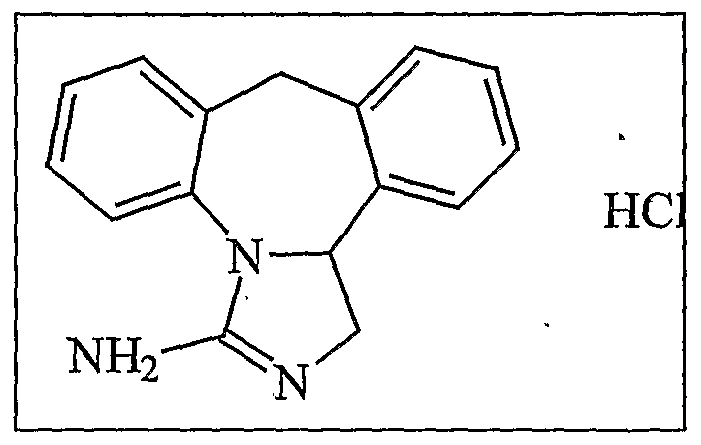
Formula (IA)
US 4313931 (herein after refers as '931 patent) discloses 3-amino-9,13b-dihydro-lH- dibenz-[c,fjimidazolo[l,5-a]-azeρine (Epinastine), non-toxic pharmacologically acceptable acid addition salt thereof and process for preparing the same. This patent also discloses pharmaceutical composition containing Epinastine or its salts and method for treating bronchial asthma and allergic bronchitis. The '931 patent does not discloses or discuss about the solid-state characterization of Epinastine base or its HCl salt. XRPD pattern of the Epinastine HCl obtained by this process is disclosed in JP 2004-300042.
US 5312916 relates to process for preparing 3-amino-9,13b-dihydro-lH-dibenz[c,fJimi- dazolo[l,5-a]-azepine hydrochloride using dimethylformamide with no disclosure about solid state properties of the isolated product of Epinastine hydrochloride.
US 6403790 relates to a process for preparing Epinastine hydrochloride in the high-melting crystal modification using water as solvent. The 790 patent also discloses that Epinastine hydrochloride exists in low melting crystal modification which melts at 250-2630C and high melting crystal modification which melts at 275-281 0C.
JP 2004-300042 discloses a crystal polymorph of Epinastine HCl5 which is characterized by XRPD. The patent also discloses the method for producing the crystal polymorph which involves crystallizing high quality epiastin hydrochloride from an aprotic polar solvent. XRPD pattern of the Epinastine HCl obtained by following the process disclosed in US 4313931 is also disclosed in this patent.
Many organic compounds including active pharmaceutical ingredients (API's) exhibits polymorphisms and can exist in many crystalline forms. Polymorphism is the ability of the compound to exhibit more than one orientation or conformation of molecule within the crystal lattice. Drug substance exists in various polymorphic forms; differ from each other in terms of their stability, solubility, compressibility, flowability and spectroscopic properties, thus affecting dissolution, bioavailability and handling characteristics of the substance.
When the difference in the solubility of various polymorphs is sufficiently large, it may alter the drug product in vivo dissolution and hence drug product bioavailability. Rate of dissolution of an API in patient's stomach fluid can have therapeutic consequences since it imposes an upper limit on the rate at which an orally administrated API can reach the patient bloodstream. Flowability affects the ease with which the material is handled while processing a pharmaceutical product. Knowledge of an existence of different crystal phases and their overall physical and chemical behaviour is required for selection of polymorphic form to be used in the preparation of final dosage form. Towards this end, investigation of crystal polymorphisms is an essential step in pharmaceutical research due
to the influence of solid-state properties on dosage form. The discovery of new polymorphs with same or better pharmaceutical equivalence and bioequivalence as that of the existing polymorphs provides an opportunity to improve the performance characteristic of the pharmaceutical product. There is disclosed in the prior art that Epinastine HCl exists in different crystal modification however, the prior art does not disclose the polymorphic forms of Epinastine base. Epinastine hydrochloride can still exist in different crystal structures other than known crystal modifications as disclosed in prior art. Based on the known crystal modifications of Epinastine HCl, there is still considered need to have different new polymorphs of Epinastine or its salt which may exhibits better solubility and dissolution rate. It has now been found, surprisingly, new crystal structures of Epinastine and its hydrochloride salt and these new polymorphs having improved bulk handling and dissolution properties can be more valuable and practical with respect, to physical and chemical stability compared to the existing crystal modifications of Epinastine.
All the polymorphic forms of Epinastine hydrochloride reported in the prior art are crystalline in nature. It is known that the amorphous form of drug substance exhibits different dissolution characteristic. Amorphous forms can have solubility, several hundred times than that of the crystalline counterparts. Thus the present invention also provides novel amorphous form of Epinastine and hydrochloride salt thereof.
Objects of the invention:
The object of the present invention is to provide novel crystalline forms, Form I and Form II of Epinastine base and process of preparation thereof.
Another object of the invention is to provide novel amorphous form of Epinastine base and process for preparation thereof.
Yet another object of the invention is to provide seven novel crystalline forms, Form II, Form III, Form IV, Form V, Form VI, Form VII and Form VIII of Epinastine HCl and process for preparing them.
Another object of the present invention is to provide novel amorphous form of Epinastine HCl and process for preparation thereof.
Still further object of the present invention is to provide pharmaceutical composition containing novel crystalline or amorphous forms of Epinastine base or its hydrochloride salt.
Another object of the present invention provides method of treatment of anti-allergic and anti-histaminergic activity with therapeutically effecting amount of the pharmaceutical composition comprising novel crystalline or amorphous forms of Epinastine base or its hydrochloride salt.
Summary of the invention:
According one aspect of the present invention novel crystalline forms, Form I and Form II and amorphous form of Epinastine base and process of preparation thereof is disclosed.
According to another aspect, the present invention provides seven crystalline forms, Form II, Form III, Form IV, Form V, Form VI, Form VII and Form VIII and amorphous form of Epinastine HCl and process for preparation thereof.
According to another aspect, the present invention provides pharmaceutical composition comprising novel crystalline or amorphous forms of Epinastine base or its hydrochloride salt.
According to another aspect, the present invention provides method of treatment of antiallergic and anti-histaminergic activity with therapeutically effecting amount of the pharmaceutical composition comprising novel crystalline or amorphous forms of Epinastine base or its hydrochloride salt.
Brief description of figures:
FIG. 1 is a characteristic X-ray Powder diffraction pattern of Epinastine base Form I. FIG. 2 is a characteristic X-ray Powder diffraction pattern of Epinastine base Form II. FIG. 3 is characteristic X-ray Powder diffraction pattern of Amorphous Epinastine Base.
FIG. 4 is a characteristic X-ray Powder diffraction pattern of Epinastine HCl Form II. FIG. 5 is a characteristic X-ray Powder diffraction pattern of Epinastine HCl Form III. FIG. 6 is a characteristic X-ray Powder diffraction pattern of Epinastine HCl Form IV. FIG. 7 is a characteristic X-ray Powder diffraction pattern of Epinastine HCl Form V. FIG. 8 is a characteristic X-ray Powder diffraction pattern of Epinastine HCl Form VI. FIG. 9 is a characteristic X-ray Powder diffraction pattern Epinastine HCl Form VII. FIG. 10 is a characteristic X-ray Powder diffraction pattern of Epinastine HCl Form VIII. FIG.11 is a characteristic X-ray Powder diffraction pattern of Amorphous Epinastine HCl.
Description of the present invention:
The present invention describes novel crystalline forms, Form I and Form II of Epinastine base and seven crystalline forms, Form II, Form III, Form IV, Form V, Form VI, Form VII and Form VIII of Epinastine HCl. In accordance with the present invention there is also provided process for preparation of crystalline forms of Epinastine base and its hydrochloride salt. The present invention further provides amorphous form of Epinastine base and its hydrochloride salt and process for preparation thereof.
According to one embodiment of the invention there is provided a novel crystalline form, Form I Epinastine base characterized by the XRPD peaks given below:
Pos. r°2Th.l ReI. Int. \%]
5.89 2.18
8.73 3.58
Ct 11.65 48.31
13.87 71.73
14.30 3.13
15.09 19.58
16.13 13.88
17.14 28.82
17.49 12.80
18.15 100
19.04 35.28
19.89 56.45
21.81 24.99
22.33 7.80
22.65 6.71
23.06 21.59
23.33 18.19
23.79 24.86
24.15 25.99
24.51 6.30
25.26 35.77
26.16 35.83
26.63 26.98
27.19 19.41
27.79 16.60
28.66 16.31
29.05 14.70
30.33 11.19
31.45 8.56
32.42 6.34
33.47 4.52
33.80 5.22
36.32 4.90
36.67 6.21
37.64 6.04
39.64 2.79
40.45 1.81
42.10 3.03
42.96 2.12
43.89 3.91
48.49 1.18
Another embodiment of the invention provides process for preparing the crystalline Epinastine base Form I comprising the steps of, a) dissolving Epinastine base in solubilizing solvent; b) cooling and stirring the obtained solution; c) isolating the separated product .
The solubilizing solvent is selected from a group comprising aliphatic ketones, nitriles and C1-C4 alcohols. The ketone is selected from the group consisting of acetone, ethyl methyl ketone, diethyl ketone preferably acetone. Nitrile used is acetonitrile and Ci-C4 alcohols are selected from group consisting of methanol, ethanol, 1- propanol, isopropyl alcohol (IPA), butanol preferably IPA.
In preferred embodiment Epinastine base is dissolved at reflux temperature of the solvent selected for dissolution or at temperature of 60-80°C. The obtained solution is cooled
followed by stirring at temperature 5-1O0C for 2-8 hours. The separated solid is then isolated by filtration followed by drying at temperature range of 30-900C preferably 650C to get Epinastine base Form I.
In another embodiment of the present invention process for preparation of crystalline Epinastine base Form I comprises the following steps; a) dissolving/suspending Epinastine base in a solubilizing solvent; b) adding anti-solvent to the obtained solution; c) isolating the separated solid.
The solubilizing solvent is selected from group comprising polar aprotic solvent, aliphatic cyclic ethers, aliphatic ketone and chlorinated hydrocarbons or mixtures thereof.
The polar aprotic solvent is selected from dimethyl formamide (DMF), dimethyl sulfoxide (DMSO), dimethyl acetamide (DMA). The aliphatic cyclic ether is selected from 1,4 - dioxane or tetrahydrofuran (THF) or mixtures thereof, preferably 1,4-dioxane. The aliphatic ketone isj acetone. The chlorinated hydrocarbon is selected from chloroform or methylene dichloride (MDC) preferably MDC.
The anti-solvent is selected from the group comprising of aliphatic ketones is selected from acetone, 2-butanone, diethyl ketone or mixtures thereof, preferably acetone, esters is selected from methyl acetate, ethyl acetate, butyl acetate or mixtures thereof, preferably ethyl acetate., hydrocarbon is selected from n-hexane and n-heptane preferably n-hexane , ethers is selected from diethyl ether (DEE), diisopropyl ether (DEPE), methyl tert-butyl ether (MTBE) preferably DIPE, aromatic hydrocarbons is selected from toluene and xylene preferably toluene and water.
In preferred embodiment of the invention Epinastine base is dissolved at reflux temperature of individual solvent selected for dissolution or at temperature 40-800C. The anti-solvent is added to the obtained solution and then stirred at temperature range of 0- 3O0C preferably 25-3O0C for several hours. The separated solid is isolated by filtration followed by drying at temperature 650C to get Epinastine base Form I.
Another embodiment of the present invention provides crystalline Epinastine base Form II characterized by the following XRPD peaks which are shown in the table below:
Pos. r°2Th/| ReI. Int. [%1
5.18 0.70
10.23 100
12.73 2.09
13.83 0.20
15.29 12.34
16.50 2.58
19.68 1.25
20.39 10.44
20.86 11.54
22.08 0.50
° 24.30 7.71
25.53 2.44
26.48 0.54
27.76 0.53
29.13 1.11
30.40 2.39
31.51 0.45
34.10 3.80
35.40 2.27
35.97 16.90
39.22 2.13
40.55 2.27
41.31 2.20
46.73 2.95
47.18 0.59
In another embodiment of the invention, process for preparation of Epinastine base Form II according to the invention comprises the following steps, a) dissolving Epinastine base in a solubilizing solvent to obtain a solution; b) pouring the solution obtained in anti-solvent; c) stirring the solution for several hours and d) isolating the product .
The solubilizing solvent is selected from group comprising alcohols, aliphatic cyclic ethers or mixtures thereof. The alcohol is selected from methanol, ethanol, 1-propanol, 2- propanol (IPA), butanol more preferably IPA. Aliphatic cyclic ether is selected from 1,4- dioxane, tetrahydrofuran preferably 1,4-dioxane.
The anti-solvent selected is any suitable solvent which is miscible with the above solubilizing solvents most preferably water. The anti-solvent is cooled to -10 to 350C preferably 0-5°C.
In preferred embodiment of the invention, Epinastine base is dissolved at reflux temperature of individual solvent selected for dissolution or at temperature 60-80°C. The obtained solution is then poured into anti-solvent at temperature of 0-100C and stirred at the same temperature for 2-4 hours. The separated solid is isolated by filtration followed by drying at 650C to get Epinastine Form II.
According to another embodiment, the present invention provides novel amorphous form of Epinastine. The amorphous Epinastine is characterized by, having broad x-ray diffraction spectrum as in figure 3.
Another embodiment of the present invention provides process for the preparation of amorphous Epinastine which comprises dissolving Epinastine base in a suitable solubilizing solvent at reflux temperature of suitable solvent selected for dissolution or at temperature range of 20-500C and the solvent is removed from the clear solution by vacuum drying or spray drying technique to get amorphous Epinastine .
The solubilizing solvent is selected from alcohol, chlorinated hydrocarbons or mixture thereof. The alcohol is selected from methanol, ethanol, preferably methanol. The chlorinated hydrocarbon is selected from methylene dichloride (MDC) or chloroform preferably methylene dichloride.
The concentration of Epinatin base is 5-15% preferably 8-12 % w/v. The spray drying is carried out at inlet temperature of 40 to 1700C, preferably 1600C and outlet temperature of 35-85°C, preferably 650C. The vacuum evaporation is carried out at temperature about 65-85°C.
Epinastine HCl obtained by following the process disclosed in US 4313931 is designated herein as 'Form F of Epinastine hydrochloride.
According to another embodiment, the present invention provides new form of Epinastine hydrochloride designated as Form II characterized by the XRPD peaks which are shown in the table below:
Pos. r°2Th.l ReI. Int. f%l
8.46 7.20
9.29 76.16
10.19 79.93
12.50 10.09
13.27 21.75
14.72 16.61
16.24 11.06
16.80 7.28
17.45 39.45
18.44 57.90
18.91 18.26
19.33 8.38
19.66 9.51
20.75 15.82
22.10 42.18
22.70 8.43
23.36 24.35
24.20 100
24.73 15.18
25.58 29.40
26.47 11.88
27.71 49.72
28.31 30.90
28.97 43.01
29.40 22.31
30.45 19.23
31.31 18.36
31.86 40.69
33.76 9.81
34.52 14.07
35.12 16.64
36.39 8.37
37.15 19.48
38.36 21.94
38.66 13.89
42.07 17.07
44.48 6.12
47.49 4.80
In another embodiment of the present invention the process for preparing Epinastine hydrochloride Form II comprises the steps of, a) suspending Epinastine base in solubilizing solvent b) adding acid to the obtained solution; c) warming the solution to get solution and d) isolating the separated solid.
Epinastine base can be of any polymorphic form used for making novel form of Epinastine HCl. The solubilizing solvent is aqueous solvent selected such that Epinastine base is insoluble but its hydrochloride salt is freely soluble, preferably water. The acid used is hydrochloric acid, preferably used in concentrated form, the term "concentrated form" meaning an approximately 32% by weight aqueous hydrochloric acid.
In preferred embodiment of the invention, Epinastine base is suspended in solubilizing solvent and acid is added to the obtained suspension .The solution is warmed at reflux temperature or at about 50-60°C to get clear solution. The clear solution is then filtered and cooled to -10 to 50C preferably 0-5°C. The suspension is further stirred at temperature 10-150C for 30 min. The separated solid is then filtered and dried at 25-3O0C to get Epinastine HCl Form II.
According to another embodiment, the present invention provides new form of Epinastine hydrochloride designated as Form III characterized by the following XRPD peaks which are shown in the table below:
Pos. [°2Th.l ReI. Int. [%1
5.62 100
8.86 27.15
11.27 37.99
12.77 5.88
13.18 16.22
13.67 22.13
14.77 16.41
15.43 20.21
16.93 21.69
17.38 19.14
17.74 37.66
18.23 9.51
18.83 13.69
20.17 9.24
20.73 3.27
21.30 29.55
21.80 13.18
22.60 13.94
23.46 21.78
23.81 16.41
24.52 21.7
25.00 27.55
25.51 8.84
25.94 10.19
26.46 17.61
27.17 6.S
27.49 9.74
28.20 31.58
29.26 5.99
29.75 9.56
30.23 8.2
32.54 11.02
33.78 5.08
34.96 3.64
39.29 4.04
40.97 4.96
42.61 2.15
46.71 2.82
Another embodiment of the present invention provides process for preparing Epinastine hydrochloride Form III comprising the steps of, a) dissolving Epinastine hydrochloride in a solubilizing solvent; b) adding suitable antisoivmt; c) isolating the product and drying
Another embodiment of the present invention provides process for preparing Epinastine hydrochloride Form III comprising the steps of, a)dissolving Epinastine hydrochloride in a solubilizing solvent; b)isolating the product and drying.
Epinastine hydrochloride selected for making novel form of Epinastine hydrochloride can be of any polymorphic forms preferably Form II.
The solubilizing solvent is selected from the group comprising of C1-Gt alcohol, chlorinated hydrocarbon and aliphatic cyclic ether. The C1-C4 alcohol is selected from the group of methanol, ethanol, 1-propanol, isopropyl alcohol, butanol preferably 2-propanol. Chlorinated hydrocarbons selected are methylene dichloride (MDC) or chloroform preferably MDC. The aliphatic cyclic ether is selected from 1,4-dioxane or tetrahydrofuran (THF).
The anti-solvent is selected from group of aliphatic acyclic ether, hydrocarbon and ester. The aliphatic acyclic ether is selected from DEE, DIPE or MTBE preferably DIPE. Hydrocarbon is selected from pentane, hexane, heptane preferably hexane and ester is selected from ethyl acetate, butyl acetate, isopropyl acetate preferably ethyl acetate. The anti-solvent is added at 3O-5O°C preferably 25-35°C. The suspension is further cooled and stirred at 5-1O0C for 2-4 hours. The separated solid is filtered and dried at 90°C to get the novel Form III of Epinastine hydrochloride.
In preferred embodiment of the invention, Epinastine hydrochloride is dissolved in suitable solubilizing solvent at reflux temperature or at temperature 50-600C. The anti- solvent is added at 30-500C preferably 25-350C. The suspension/solution is further stirred at 5-100C for 2-4 hours. The separated solid is filtered and dried at 9O0C to get the novel Form III of Epinastine hydrochloride.
According to another embodiment the present invention provides new form of Epinastine hydrochloride designated as Form IV characterized by the following XRPD peaks which are shown in the table below:

9.22 22.02
9.86 37.27
10.36 16.13
10.95 12.21
11.59 32.27
11.79 9.92
12.37 8.09
12.89 12.09
13.18 15.73
13.41 12.48
15.57 14.67
16.91 15.80
17.37 17.76
18.39 35.81
18.90 27.13
19.86 23.12
20.78 100
21.13 48.51
21.70 20.52
22.57 18.74
23.16 53.2
24.48 20.46
25.48 17.38
25.92 27.09
26.45 29.72
27.56 24.98
28.58 22.02
29.14 20.39
30.43 15.04
31.92 9.48
32.98 8.37
34.05 6.20
36.44 1.61
In another embodiment of the present invention the process for preparing Epinastine hydrochloride Form IV comprises the steps of, a) dissolving Epinastine HCl in a solubilizing solvent; b) adding antisolvent to the obtained solution; c) isolating the product and drying.
Epinastine hydrochloride selected for making novel form of Epinastine hydrochloride can be of any polymorphic form, preferably Form II.
The solubilizing solvent is selected ftom the group of C1-C4 alcohol or 1,4-cyclohexane. The alcohol is selected are methanol, ethanol, 2-propanol, preferably methanol.
The antisolvent is selected from group of aliphatic acyclic ether, aromatic hydrocarbon and ester or mixtures thereof. The aliphatic acyclic ether is selected from DEE, DIPE or MTBE preferably DIPE. Aromatic hydrocarbon is selected from toluene, xylene
preferably toluene and ester is selected from ethyl acetate, butyl acetate, iso propyl acetate preferably ethyl acetate.
The anti-solvent is added at 30-600C preferably 25-35°C. The suspension is further stirred at 5-10°C for 2-4 hours. The separated solid is filtered and dried at 50-65°C to get the novel form, Form IV of Epinastine hydrochloride.
In preferred embodiment of the invention Epinastine hydrochloride is dissolved in suitable solubilizing solvent at reflux temperature or at temperature 50-60°C. The anti- solvent is added at 30-500C preferably 25-350C. The suspension is further stirred at 5-1O0C for 2-4 hours. The separated solid is filtered and dried at 50-650C to get the novel Epinastine hydrochloride Form IV .
According to another embodiment the present invention provides new form of Epinastine hydrochloride, designated as Form V characterized by the following XRPD peaks which are shown in the table below:
Pos. r°2Th.l ReI. Int. \%\
5.81 5.81
8.62 10.45
10.32 32.43
11.12 7.92
11.57 26.42
12.79 21.28
13.13 21.99
14.06 5.22
14.37 6.89
14.91 5.62
15.48 30.88
17.29 38.18
18.09 5.50
19.38 19.10
19.86 31.74
20.53 18.80
20.81 100
21.08 19.07
21.43 22.36
21.63 32.21
22.24 17.69
22.89 10.62
23.90 14.73
24.39 12.77
24.81 15.20
25.88 20.22
26.23 11.69
27.21 13.15
27.80 30.98
28.19 14.56
28.47 26.66
29.03 14.16
30.58 5.06
31.19 9.36
31.81 6.56
32.43 8.43
34.97 4.45
36.56 4.18
37.49 2.20
40.33 2.18
41.24 2.69
43.21 2.19
In another embodiment of the present invention the process for preparing Epinastine hydrochloride Form V comprises the steps of, a) dissolving Epinastine HCl in solubilizing solvent; b) adding anti-solvent to the obtained solution; c) stirring the suspension for several hours; d) isolating the product and drying.
According to another embodiment the present invention provides process for preparation of Epinastine hydrochloride Form V which comprises suspending Epinastine hyrochloride (in any form) in suitable solubilizing solvent and isolating the product.
Epinastine hydrochloride selected for making novel form of Epinastine hydrochloride can be of any polymorphic forms preferably Form II.
The solubilizing solvent is selected from C1-C4 alcohol such as methanol, ethanol, 2- propanol, preferably ethanol and aromatic hydrocarbon such as toluene, xylene preferably toluene. The anti-solvent is selected from a group of aliphatic acyclic ether such as DEE, DIPE or MTBE. The anti-solvent is cooled to -10 to 35°C preferably 10-25°C.
In preferred embodiment of the invention, Epinastine hydrochloride is dissolved in suitable solubilizing solvent at reflux temperature or at temperature of 50-600C. The anti- solvent is added at 30-50°C preferably 25-35°C. The suspension is further cooled and stirred at 5-l0°C for 2-4 hours. The separated solid is filtered and dried at 50-650C to get the novel form, Epinastine hydrochloride Form V.
According to another embodiment the present invention provides new form of Epinastine hydrochloride designated as Form VI characterized by the following XRPD peaks which are shown in the following table below,

11.17 3.2
13.27 2.62
14.25 16.23
16.87 32.00
18.00 18.97
18.17 13.57
18.55 3.17
19.65 7.41
20.34 63.81
20.60 26.48
20.99 17.93
22.21 69.24
22.80 8.94
24.02 52.60
24.91 38.53
25.83 16.76
26.57 24.48
27.11 33.27
27.48 16.74
28.96 17.40
29.42 15.26
30.14 21.31
31.01 21.34
31.44 16.39
32.65 5.73
33.72 6.53
34.32 5.40
34.94 8.54
36.47 6.85
37.69 9.20
39.25 5.15
40.20 5.18
40.73 4.16
43.31 6.07
44.07 5.61
44.67 5.31
45.13 6.66
47.51 7.92
49.14 5.12
In another embodiment of the present invention the process for preparing Epinastine hydrochloride Form VI comprises the steps of, a) dissolving Epinastine HCl in solubilizing solvent; b) adding antisolvent to the obtained solution; c) isolating the product and drying.
Epinastine hydrochloride selected for making novel form of Epinastine hydrochloride can be of any polymorphic forms preferably Form II.
The solubilizing solvent is selected from the group of C1-C4 alcohols, cyclic ethers and chlorinated hydrocarbons. The alcohol selected are methanol, ethanol, 2-propanol, preferably ethanol. The aliphatic cyclic ether selected are 1,4-dioxane, THF preferably 1,4-dioχane and chlorinated hydrocarbon selected are MDC, chloroform preferably MDC.
The anti-solvent is selected from a group of aliphatic acyclic ethers or hydrocarbons. The aliphatic acyclic ether is selected from DEE5 DIPE or MTBE and aliphatic hydrocarbon is selected from pentane, hexane or heptane preferably n-hexane.
In preferred embodiment of the invention Epinastine hydrochloride is dissolved in suitable solubilizing solvent at reflux temperature or at temperature 50-60°C. The anti- solvent is added at 30-50°C preferably 25-350C. The suspension is further stirred at 5-1O0C for 2-4 hours. The separated solid is filtered and dried at 50-650C to get the novel Form VI of Epinastine hydrochloride.
According to another embodiment the present invention provides new form of Epinastine hydrochloride designated as Form VII characterized by the following XRPD peaks which are shown in the table below,
Pos. r°2Th.l ReI. Int. \%\
5.91 26.86
8.62 13.05
10.39 49.08
11.12 4.77
11.57 30.85
11.77 18.25
12.83 17.15
13.15 17.33
13.76 5.96
14.43 11.11
14.97 8.82
15.28 17.82
15.49 27.43
17.22 20.97
17.66 9.88
18.20 9.97
18.95 4.25
19.35 26.84
19.92 26.53
20.53 18.84
20.75 100
21.11 14.10
21.60 47.40
21.86 17.39
22.44 19.22
22.91 12.86
23.96 12.74
24.19 13.46
24.51 19.85
25.49 14.70
25.76 7.33
25.94 23.79
26.39 19.37
27.38 24.34
27.83 35.16
28.46 31.47
29.10 10.81
29.95 10.94
31.36 10.47
31.90 8.34
In another embodiment of the present invention the process for preparing Epinastine hydrochloride Form VII comprises the steps of, a) dissolving Epinastine HCl in a solubilizing solvent;
b) adding anti-solvent to the obtained solution; c) stirring the solution for several hours; d) isolating the product and drying
Epinastine HCl selected for making novel form of Epinastine HCl can be of any polymorphic forms preferably Form II.
The solubilizing solvent is selected from the group of cyclic ether and polar aprotic solvent. The aliphatic cyclic ether is selected from 1,4-dioxane, tetrahydrofuran (THF) preferably 1,4-dioxane and polar aprotic solvent is selected from dimethyl formamide (DMF), dimethyl sulfoxide (DMSO), N,N-dimethyI acetamide (DMA) preferably DMF.
The anti-solvent is selected from a group of aliphatic ketones and esters. The aliphatic ketone is selected from acetone, 2-butanone, diethyl ketone preferably acetone and ester is selected from ethyl acetate, butyl acetate, iso propyl acetate, preferably ethyl acetate.
In preferred embodiment of the invention Epinastine hydrochloride is dissolved in suitable solubilizing solvent at reflux temperature or at temperature 50-60°C. The anti- solvent is added at 30-500C preferably 25-350C. The suspension is further stirred at 5-100C for 2-4 hours. The separated solid is filtered and dried at 50-650C to get the novel Form VII of Epinastine hydrochloride.
According to another embodiment the present invention provides new form of Epinastine hydrochloride designated as Form VIII characterized by the following XRPD peaks which are shown in the table below,
Pos. r°2Th.l ReI. Int. [%1
5.94 88.32
9.08 7.13
11.79 61.54
13.81 27.19
14.22 24.28
14.99 17
15.29 100
15.51 26.74
17.64 29.85
18.30 36.83
18.99 13.08
19.60 73.77
21.53 67.58
21.89 78.32
22.19 23.49
22.49 50.62
22.95 11.91
24.20 80.71
24.98 52.74
25.52 55.23
25.82 24.36
26.47 63.35
27.09 24.21
27.40 89.21
28.79 31.37
29.97 41.99
30.26 37.71
30.46 30.49
31.09 9.66
31.85 11.93
32.70 10.41
33.03 16.80
35.00 11.51
35.56 9.95
36.70 9.79
37.59 6.23
38.84 7.82
39.53 10.27
40.41 18.00
43.26 8.00
In another embodiment of the present invention the process for preparing Epinastine hydrochloride Form VIII comprises the steps of, a) dissolving Epinastine HCl in a solubilizing solvent b) adding antisolvent to the obtained solution; c) stirring the suspension for several hours. d) isolating the product and drying
According to yet another embodiment the process for preparation of Epinsatine HCl Form VIII comprises dissolving Epinastine HCl in a solubilizing solvent and stirring the obtained solution at 5-1O0C for several hours preferably 2-6 hours to get the solid of Epinsatine HCl Form VIII.
In yet another embodiment of the present invention the process for preparing Epinastine hydrochloride Form VIII comprises the steps of, e) dissolving Epinastine HCl in a solubilizing solvent f) pouring the obtained solution to anti-solvent ; g) stirring the suspension for several hours, h) isolating the product and drying
Epinastine HCl selected for making novel form of Epinastine HCl can be of any polymorphic forms preferably Form II.
The solubilizing solvent is selected from the group of chlorinated hydrocarbon, aliphatic cyclic ethers and nitrile. The chlorinated hydrocarbon is selected from methylenedichloride (MDC), chloroform preferably MDC, aliphatic cyclic ethers selected are 1,4-dioxane, THF preferably 1,4-dioxane and nitrile used is acetonitrile.
The anti-solvent is selected from group of aliphatic ketone, esters and ether. The aliphatic ketone is selected from acetone, 2-butanone, diethyl ketone preferably acetone, ester selected is ethyl acetate, butyl acetate preferably ethyl acetate and ether is selected from DEE, DIPE and MTBE. The anti-solvent is cooled to -10 to 300C preferably 20-25°C.
In preferred embodiment of the invention Epinastine hydrochloride is dissolved in suitable solubilizing solvent at reflux temperature or at temperature 50-60°C to obtain the solution. The obtained solution is added to the cooled antisolvent or anti-solvent is added to the obtained solution at 30-50°C preferably 25-350C. The suspension is further stirred at 5-100C for 2-4 hours. The separated solid is filtered and dried at 50-650C to get the novel Form VIII of Epinastine hydrochloride.
According to another embodiment the present invention provides a novel amorphous form of Epinastine HCl. The amorphous Epinastine hydrochloride in accordance with the present invention is characterized by having broad x-ray diffraction spectrum as in figure 11.
Another embodiment of the present invention provides process for the preparation of amorphous Epinastine HCl which comprises dissolving Epinastine HCl in a suitable solubilizing solvent at a temperature range of 20-400C and the solvent is removed from the obtained solution by vacuum drying or spray drying or lyophilization technique to get amorphous Epinastine HCl .
The solubilizing solvent is selected from water, alcohol or mixtures thereof preferably water. The alcohol is selected from methanol and ethanol preferably methanol. The concentration of Epinastine HCl in solution is 5-15% preferably 8-12 %. The spray dried is carried out at inlet temperature of 40 to 170°C, preferably 160°C and outlet temperature of 35-850C, preferably 650C. The vacuum evaporation is carried out at temperature ranges from 65-85°C The lyophilization is carried out by freeze drying an aqueous solution at temperature -20 to -8O0C under vacuum preferably -400C.
As used herein, the phrase room temperature refers to a temperature at about 25° C.
The crystallization process hitherto described to prepare the novel polymorphs consists of dissolving Epinastine base or Epinastine HCl in the selected solvent either with or without heating, preferably with heating at or near boiling point of the solvent. Cooling the resultant solution to -100C to 5°C for several hours to regenerate the solid. Isolating the precipitated solids and drying the isolated solids at about ambient to 650C temperature.
The solvent and anti-solvent combination process described to prepare the novel polymorphs consist of dissolving Epinastine base or Epinastine HCl in the suitable solvent. The dissolution may be carried out at room temperature or under reflux condition. Adding anti-solvent to the resulting solution under warm condition to get polymorphs of Epinastine base or Epinastine HCl. The anti-solvent addition is carried out at room temperature or at temperature of 25-350C. Isolating the precipitated solids by filtration and drying the isolated solids at about ambient temperature to 65°C or at room temperature. The novel polymorphs of Epinastine base and Epinastine HCl in accordance with the present invention are characterized by X-ray powder diffraction. X-ray powder diffraction pattern has been obtained on Xpert'PRO, Panalytical, diffractometer equipped
with accelerator detector using Copper Ka (λ = 1.5406 A) radiation with scanning range between 4-50 θ at scanning speed of 27min.
The following examples illustrate the invention described above, however they are not intended to limit the scope of the invention in any manner.
EXAMPLES Example A:
Preparation of 6-aminometkyl-6,ll-dihydro-5H-dibenzo[b,e]azepine:
A solution of aluminum hydride in tetrahydrofuran was produced by slowly adding drop wise solution of 275 gm of 98% sulfuric acid in 0,6 liters of anhydrous tetrahydrofuran to a suspension of 216 gm of lithium aluminum hydride in 7.5 liters of absolute tetrahydrofuran under stirring. Without separating Lithium sulphate that forms, a solution of 300 gm of 6-Cyano azepine in 2.4 liters tetrahydrofuran was added within 30 min. The reaction mixture was stirred for 2 hours at room temperature and then decomposed while maintaining temperature below 0 0C of the hydride by addition of 0.75 liters of water. The inorganic salts were removed by suction filtration. The filtrate was evaporated under reduced pressure at 50-55°C completely to get dark reddish brown colored oil.
Example B:
The above prepared amino compound was dissolved in 1.5 L methanol at temperature 25-30 0C . 112 gm of fumaric acid was added lot wise in 30-40 minutes at 35-400C and the reaction mixture was stirred for 1 hour. The solvent was removed completely under vacuum at temperature below 600C . 1.5 L acetonitrile was added to the obtained reaction mass and stirred for 25 to 30 minutes at temperature 25-300C . the reaction mixture was filtered and suck dried. The wet cake of fumarate salt was suspended in 1.12 L water and sodium hydroxide solution (87 gm NaOH dissolved in 450 ml water) was added slowly at temperature 25-300C followed by stirring for 30 minutes. The obtained Epinastine amine was extracted with 2x2.25 L MDC. The MDC layer was washed with water, dried on sodium sulphate and concentrated to get pure Epinastine amine. Yield: 200-205 gm (64-65% of Theory).
Example C: Preparation of 3-amino-9,13b-dihydro-lH-dibenzo[c,f]iinidazo[l,5-a] azepine hydrobromide: 365gm of 6-aminomethyl-6,ll-dihydro-5H-dibenzo[b,e]azepine was dissolved in 3.65 liters ethanol and a solution of 176 gm cyanogen bromide in 1.5 liters of absolute tetrahydrofuran was added drop wise under stirring at room temperature. The reaction mixture was stirred overnight at room temperature and then admixed with 2.74 liters of diethyl ether. Subsequently, resulted crystals were filtered and washed with 0.70 liters of diethyl ether.
Yield: 340-345 Gms (63-65% of Theory)
Example D: Purification of Epinastine hydro bromide
0.20 Kg of Epinastine hydro bromide was added to 2 liters of dimethyl formamide and heated to at 80 0C to get the clear solution. The reaction temperature was maintained for one hour at this temperature and then cooled the reaction up to ambient temperature. 2.0 liters of ethyl acetate was admixed to the obtained reaction mixture and resulted crystals were filtered and washed with 0.50 liters of ethyl acetate. Yield: 185-190 Gms. (93-95% of Theory)
Example ErPreparation of 3-amino-9,13b-dihydro-lH-dibenzo[c,fjiinidazo[l,5-a] azepine (Epinastine Base): 180 gm of 3-amino-9,13b-dihydro-lH- dibenzo[c,fjimidazo[l,5-a] azepine hydro bromide was admixed with 500 ml of 5% sodium hydroxide solution. The reaction mixture was stirred for 3 hours at room temperature maintaining the pH of the solution more than 10 on pH measuring paper strip. After stirring for 3 hours, filter the solution at ambient temperature and washed with 250 ml of water twice. pH of the filtrate should not more than 7 to get the solid which was dried at 40 0C overnight under vacuum till desired LOD achieved. Yield: 125-130 gm (92-96% of Theory)
Example F: Preparation of 3-amino-9,13b-dihydro-lH-dibenzo[c,f]imidazo[l,5-a] azepine HCl Form III: 100 gm of 3-ammo-9,13b-dihydro-lH-dibenzo[c,fJimidazo[l,5- a] azepine was suspended in 0.3 liters 1,4-dioxane and to this 41.8 ml cone. HCl was added. The clear solution obtained was rapidly added to 6.1 liters ethyl acetate followed by stirring for 30 minutes, filtered the solution at ambient temperature to isolate Epinastine HCl Form III. Yield: 108 gm (92-96%)
Example G: Conversion of Form III of 3-amino-9,13b-dihydro-lH- dibenzo[c,fjimidazo[l,5-a] azepine HCI to Form VIII: 100 gm of 3-amino-9,13b- dihydro-lH-dibenzo[c,f]iinidazo[l,5-a] azepine HCl Form III was suspended in 1 liters acetonitrile and heated to reflux for 24 hrs under stirring to ensure complete transformation of Form III to Form VIII. The solution obtained was filtered at ambient temperature to isolate Epinastine HCl Form VIII. Epinastine HCl Form VIII obtained was suspended in 1 liter ethyl acetate and heated to reflux for 24 hrs under stirring to ensure concentration of acetonitrile below 400 ppm. The solution was filtered at ambient temperature to isolate Epinastine HCl Form VIII.
Yield: 94 Gms. (92-96%)
Novel polymorphs of Epinastine or salts thereof. Epinastine Base - Form I
Example 1
0.5g of Epinastine base was dissolved in 10 ml isopropyl alcohol at reflux. The solution was filtered to remove any insoluble material. The solution was cooled and stirred at 5-1O0C for 3 hrs. The separated solid was isolated by filtration and dried at 65°C to get Epinastine Form I.
Example 2
0.5g of Epinastine base was dissolved in 60 ml acetonitrile at reflux. The solution was filtered to remove any insoluble material. The solution was cooled and stirred at 5-10°C for 3 hrs. The separated solid was isolated by filtration and dried at 65 °C to get Epinastine Form I.
Example 3
0.5 g of Epinastine base was dissolved in 60 ml acetone at reflux. The solution was filtered to remove any insoluble material. To this solution 120 ml n-hexane was added dropwise. The solution was cooled to 0-5°C. The solution was stirred for 4-8 hrs at the same temperature. The solid separated was isolated by filtration and dried at 650C to get Form Epinastine Form I.
Example 4
0.5g of Epinastine base was dissolved in 6 ml dimethylsulphoxide (DMSO) at 55-60°C. The solution was filtered to remove any insoluble material. To this solution 30 ml acetone was added dropwise. The solution was stirred at 25-30°C for 3 hrs. The separated solid was isolated by filtration and dried at 65°C to get Epinastine Form I.
Example 5
0.5g of Epinastine base was dissolved in 6 ml DMSO at 55-6O0C. The solution was filtered to remove any insoluble material. To this solution 20 ml water was added dropwise. The solution was stirred at 25-300C for 3 Hrs. The separated solid was isolated by filtration and dried at 65°C to get Epinastine Form I.
Example 6
0.5g of Epinastine base was dissolved in 10 ml methylenedichloride (MDC) at 40-450C. The solution was filtered to remove any insoluble material. To this solution 30 ml DIPE was added dropwise. The solution was stirred at 25-300C for 3 Hrs. The separated solid was isolated by filtration and dried at 650C to get Epinastine Form I.
Example 7
0.5g of Epinastine base was dissolved in 10 ml MDC at 40-450C. The solution was filtered to remove any insoluble material. To this solution 30 ml hexane was added dropwise. The solution was stirred at 25-3O0C for 3 Hrs. The separated solid was isolated by filtration and dried at 650C to get Epinastine Form I.
Example 8
0.5g of Epinastine base was dissolved in 10 ml MDC at 40-450C. The solution was filtered to remove any insoluble material. To this solution 50 ml Toluene was added dropwise. The solution was stirred at 25-3O0C for 3 Hrs. The separated solid was isolated by filtration and dried at 65°C to get Epinastine Form I.
Example 9
0.5g of Epinastine base was dissolved in 10 ml MDC at 40-450C. The solution was filtered to remove any insoluble material. To this solution 30 ml ethyl acetate was added
dropwise. The solution was stirred at 25-300C for 3 Hrs. The separated solid was isolated by filtration and dried at 65°C to get Epinastine Form I.
Example 10
0.5g of Epinastine was base dissolved in 6 ml 1,4-dioxane at reflux. The solution was filtered to remove any insoluble material. To this solution 30 ml toluene was added dropwise. The solution was stirred at 5-10°C for 3 Hrs. The separated solid was isolated by filtration and dried at 650C to get Epinastine Form I.
Example 11
0.5g of Epinastine base was dissolved in 6 ml 1,4-dioxane at reflux. The solution was filtered to remove any insoluble material. To this solution 40 ml acetone was added dropwise. The solution was stirred at 25-3O0C for 3 Hrs. The separated solid was isolated by filtration and dried at 65°C to get Epinastine Form I.
Example 12
0.5g of Epinastine base was dissolved in 6 ml 1,4-dioxane at reflux. The solution was filtered to remove any insoluble material. To this solution 25 ml hexane was added dropwise. The solution was stirred at 25-3O0C for 3 Hrs. The separated solid was isolated by filtration and dried at 65°C to get Epinastine Form I.
Example 13
0.5g of Epinastine base was dissolved in 6 ml 1,4-dioxane at reflux. The solution was filtered to remove any insoluble material. To this solution 35 ml ethyl acetate was added dropwise. The solution was stirred at 0-50C for 3 Hrs. The separated solid was isolated by filtration and dried at 65°C to get Epinastine Form I.
Example 14
0.5g of Epinastine base was dissolved in 12 ml tetrahydrofuran (THF) at reflux. The solution was filtered to remove any insoluble material. To this clear solution 30 ml water was added. The solution was stirred at 5-1O0C for 3 Hrs. The separated solid was isolated by filtration and dried at 65°C to get Epinastine Form I.
Example 15
0.5g of Epinastine base was dissolved in 12 ml THF at reflux. The solution was filtered to remove any insoluble material. To this solution 30 ml hexane was added dropwise. The solution was stirred at 25-3O0C for 3 Hrs. The separated solid was isolated by filtration and dried at 65°C to get Epinastine Form I.
Example 16
0.5g of Epinastine base was dissolved in 12 ml THF at reflux. The solution was filtered to remove any insoluble material. To this solution 30 ml DIPE was added dropwise. The solution was stirred at 25-300C for 3 Hrs. The separated solid was isolated by filtration and dried at 65°C to get Epinastine Form I.
Epinastine Base - Form II
Example 17
0.5g of Epinastine base was dissolved in 10 ml isopropylalcohol at reflux. The solution was filtered to remove any insoluble material. The clear solution was poured in 30 ml water at 0-50C. The solution was stirred at 0-50C for 3 Hrs. The separated solid was isolated by filtration and dried at 65°C to get Epinastine Form II.
Example 18
0.5g of Epinastine base was dissolved in 6 ml 1,4-dioxane at reflux. The solution was filtered to remove any insoluble material. The clear solution was poured in 30 ml water at 0-50C. The solution was stirred at 0-50C for 3 Hrs. The separated solid was isolated by filtration and dried at 650C to get Epinastine Form II.
Epinastine Base - Amorphous Form
Example 19: 5 g Epinastine base was dissolved in 10 ml methanol at a temperature range of 40-50 0C. The clear solution obtained was concentrated under vacuum at 650C to get amorphous Epinastine.
Example 20
5 g Epinastine base was dissolved in 10 ml MDC at a temperature range of 40-50 0C. The clear solution obtained was concentrated under vacuum at 650C to get amorphous
Epinastine.
Example 21
Epinastine base was dissolved in methanol at a temperature range of 30-40 0C. Concentration of Epinastine used for spray drying was about 10 % weight/volume. Spray drying is carried out at the inlet temperature 120° C and outlet temperature 650C to get amorphous Epinastine.
Example 22
Epinastine base was dissolved in MDC at a temperature range of 30-40 0C. Concentration of Epinastine used for spray drying was about 10 % weight/volume. Spray drying is carried out at the inlet temperature 120° C and outlet temperature 65°C to get amorphous
Epinastine.
Epinastine. HCl - Form II
Example 23
1 Ig Epinastine Base was suspended in 15 ml water at 30-40 0C. To this suspension 4 ml concentrated HCl (35.4%) was added dropwise. The solution was warmed to 50-60° to dissolve the solid. The solution is filtered and cooled to 0-5°C. The separated solid was further cooled to 10-15°C for 30 minutes. Filter the separated solid and dry at 25-3O0C to get Epinastine.HCl Form II.
Epinastine. HCl - Form III
Example 24.
0.5g Epinastine HCl was dissolved in 10 ml IPA at reflux temperature. To this clear solution 50 ml DIPE was added and the solution was stirred at 5-10°C for 3 hrs. the separated solid was isolated by filtration and dried at 650C to get Epinastine Form III.
Example 25
0.5g Epinastine HCl was dissolved in 10 ml IPA at reflux temperature. To this clear solution 50 ml hexane was added and the solution was stirred at 5-10°C for 3 hrs. The separated solid was isolated by filtration and dried at 650C to get Epinastine Form III.
Example 26
0.5g Epinastine HCl was dissolved in 10 ml MDC at reflux temperature. To this clear solution 30 ml DIPE was added and the solution was stirred at 5-10°C for 3 hrs. The separated solid was isolated by filtration and dried at 65°C to get Epinastine Form III.
Example 27
0.5g Epinastine HCl was dissolved in 15 ml MDC at reflux temperature. To this clear solution 30 ml ethyl acetate was added and the solution was stirred at 5-10°C for 3 Hrs. Separated solid was isolated by filtration and dried at 65°C to get Epinastine Form III.
Example 28
0.5g Epinastine HCI was dissolved in 15 ml MDC at reflux temperature. The obtained solution was cooled and stirred at 5-100C for 3 hrs. The separated solid was isolated by filtration and dried at 65°C to get Epinastine Form III.
Example 29
0.5g Epinastine HCl was dissolved in 6 ml 1,4-dioxane at 50-60°. To this clear solution 20 ml ethyl acetate was added and the solution was stirred at 5-100C for 3 hrs. The separated solid was isolated by filtration and dried at 650C to get Epinastine Form III.
Example 30
0.5g Epinastine HCl was suspended in 30 ml THF at reflux temperature. The solution was cooled and stirred at 5-100C for 3 hrs. The separated solid was isolated by filtration and dried at 65°C to get Epinastine Form III.
Epinastine. HCl - Form IV
Example 31
0.5g Epinastine HCl was dissolved in 5 ml methanol at 50-60°. To this clear solution 30 ml DIPE was added and the solution was stirred at 5-10°C for 3 hrs. The separated solid was isolated by filtration and dried at 65°C to get Epinastine Form IV.
Example 32
0.5g Epinastine HCl was dissolved in 5 ml methanol at 50-60°. To this clear solution mixture of 40 ml DIPE and 40 ml toluene was added and the solution was stirred at 5-10°C for 3 hrs. The separated solid was isolated by filtration and dried at 650C to get Epinastine Form IV.
Epinastine. HCI - Form V
Example 33
0.5g Epinastine HCl was dissolved in 10 ml ethanol at 50-60°C. To this clear solution 40 ml DIPE was added and the solution was stirred at 5-10°C for 3 hrs. The separated solid was isolated by filtration and dried at 65°C to get Epinastine Form V.
Example 34
0.5g Epinastine HCl was suspended in 30 ml toluene at reflux temperature. The solution was cooled and stirred at 5-10°C for 3 hrs. The separated solid was isolated by filtration and dried at 65°C to get Epinastine Form V.
Epinastine. HCl - Form VI
Example 35
0.5g Epinastine HCl was dissolved in 10 ml ethanol at reflux. To this clear solution 60 ml hexane was added and the solution was stirred at 5-10°C for 3 hrs and the separated solid was isolated by filtration and dried at 650C to get Epinastine Form VI.
Example 36
0.5g Epinastine HCl was dissolved in 15 ml MDC at reflux. To this clear solution 60 ml hexane was added and the solution was stirred at 5-100C for 3 hrs. The separated solid was isolated by filtration and dried at 650C to get Epinastine Form VI.
Example 37
0.5g Epinastine HCl was dissolved in 6 ml 1,4-dioxane at 50-60°. To this clear solution add 30 ml DIPE. The solution was stirred at 5-10°C for 3 Hrs. Separated solid was isolated by filtration and dried at 650C to get Epinastine Form VI.
Epinastine. HCI - Form VII
Example 38
0.5g Epinastine HCl was dissolved in 6 ml 1,4-dioxane at 50-60°. To this clear solution add 30 ml Acetone. The solution was stirred at 5-10°C for 3 Hrs. Separated solid was isolated by filtration and dried at 650C to get Epinastine Form VII.
Example 39
0.5g Epinastine HCl was dissolved in 4 ml dimethylformamide (DMF) at 50-60°. To this clear solution add 20 ml Ethyl acetate. The solution was stirred at 5-1O0C for 3 Hrs. Separated solid was isolated by filtration and dried at 650C to get Epinastine Form VII.
Epinastine. HCl - Form VIII
Example 40
0.5g Epinastine HCl was dissolved in 15 ml MDC at reflux. To this clear solution add 30 ml Acetone. The solution was stirred at 5-10°C for 3 Hrs. Separated solid was isolated by filtration and dried at 65°C to get Epinastine Form VIII.
Example 41
0.5g Epinastine HCl was dissolved in 10 ml acetonitrile at reflux! From the clear solution solid separates out at reflux. The solution was stirred at 5-10°C for 3 Hrs. Separated solid was isolated by filtration and dried at 65°C to get Epinastine Form VIII.
Example 42
0.5g Epinastine HCl was dissolved in 10 ml acetonitrile at reflux. From the clear solution solid separates out at reflux. Filter the separated solid and to the filtrate add 30 ml DIPE. The solution was stirred at 5-100C for 3 Hrs. Separated solid was isolated by filtration and dried at 65°C to get Epinastine Form VIII.
Example 43
0.5g Epinastine HCl was dissolved in 10 ml 1,4-dioxane at 50-60°. The clear solution obtained is added to 40 ml Ethyl acetate. The solution was stirred at 5-1O0C for 3 Hrs. Separated solid was isolated by filtration and dried ait 650C to get Epinastine Form VIII.
Epinastine HCl - Amorphous Form
Example 44
5 g Epinastine HCl was dissolved in 10 ml water at a temperature range of 40-50 0C. The clear solution is concentrated under vacuum at 85°C to get Amorphous Epinastine HCl.
Example 45
5 g Epinastine HCl was dissolved in 20 ml methanol at a temperature range of 40-50 0C.
The clear solution was concentrated under vacuum at 65°C to get Amorphous Epinastine
HCl
Example 46
Epinastine HCl was dissolved in water at a temperature range of 30-40 0C. Concentration of Epinastine used for spray drying is about 10 % weight/volume. Spray drying was carried out at the inlet temperature 140° C and outlet temperature 650C to get Amorphous Epinastine HCl.
Example 47
Epinastine HCl was dissolved in methanol at a temperature range of 30-40 0C. Concentration of Epinastine used for spray drying is about 10 % weight/volume. Spray drying was carried out at the inlet temperature 120° C and outlet temperature 65°C to get Amorphous Epinastine HCl.
Example 48
1Og Epinastine HCl was dissolved in 100 ml water at a temperature range of 40-50 0C. The clear solution was subjected to lyophilization for 24-48 hrs. to get Amorphous Epinastine HCl.
Claims
1. Crystalline 3-amino-9513b-dihydro-lH-dibenz-[c,f]imidazo[l,5-a]-azepine (Epinastine) having a physical characteristic selected from group consisting of: a powder X-ray diffraction pattern having peaks at about 2 θ : 5.89, 8.73, 11.65,
13.87, 15.09, 16.13, 17.14, 17.49, 18.15, 19.04, 19.89, 21.81, 23.06, 23.33, 23.79,
24.15, 25.26, 26.16, 26.63, 27.19, 27.79, 28.66, 29.05, 30.33 ± 0.2 deg.(Form I); or a powder X-ray diffraction pattern having peaks at about 2 θ : 5.18, 10.23, 12.73,
15.29, 16.50, 19.68, 20.39, 20.86, 24.30, 25.53, 29.13,30.40, 34.10, 35.40, 35.97,
39.22, 40.55, 41.31, 46.73 ± 0.2 deg. (Form II)
2. A process for preparation of Epinastine Form I comprising the steps of; a) dissolving Epinastine base in solubilizing solvent selected from the group consisting of aliphatic ketones, nitriles or Ci-C4 alcohols; and b) isolating Epinastine Form I;
3. The process as claimed in claim 2 wherein the dissolution is carried out at 550C to 65°C or at reflux temperature and isolation is carried out by cooling the solution at 0 to 3O0C with stirring for 2-8 hours.
4. A process for preparation of Epinastine Form I comprising the steps of; a) suspending Epinastine in organic solvent selected from the group consisting of polar aprotic solvent, aliphatic cyclic ether, chlorinated hydrocarbon or mixtures thereof; b) adding anti-solvent selected from the group consisting of aliphatic ketones, esters, ethers, aliphatic or aromatic hydrocarbons or mixtures thereof; and c) isolating Epinastine Form I.
5. The process as claimed in claim 4 wherein dissolution is carried out at 550C to 650C or at reflux temperature of the solvent selected for dissolution.
6. A process for preparation of Epinastine Form II comprising the steps of; a) dissolving Epinastine in solubilizing solvent selected from the group consisting of alcohols, aliphatic cyclic ethers or mixtures thereof; b) pouring the solution in water; and c) isolating Epinastine Form II
7. Epinastine having cbo between 25-400μ.
8. Amorphous 3-amino-9,13b-dihydro-lH-dibenz-[c,f]imidazolo[l,5-a]-azepine (Epinastine).
9. A process for preparation of amorphous Epinastine comprising a) dissolving Epinastine in suitable solvent selected from methanol, ethanol, methylene dichloride or chloroform; and b) spray drying the resultant clear solution or evaporating the solvent under vacuum.
10. The process as claimed in claim 9, wherein the dissolution is carried out at temperature of 20-400C, spray drying is carried out at inlet temperature of 40 to 140°C and outlet temperature of 35-850C and evaporation under vacuum is carried out at 65-850C.
11. A novel crystalline Form of 3-amino-9,13b-dihydro-lH-dibenz- [c,f]imidazolo[l,5-a]-azepine hydrochloride (Epinastine HCl) having a physical characteristic selected from group consisting of: a powder X-ray diffraction pattern having peaks at about 2 θ : 9.29, 10.19, 17.45,
18.44, 24.20, 27.71 and 31.86 ± 0.2 deg. [Form II];or a powder X-ray diffraction pattern having peaks at about 2 θ :12.77, 13.18, 13.67,
14.77, 15.43, 16.93 and 20.17 ± 0.2 deg. [Form III]; or a powder X-ray diffraction pattern having peaks at about 2 θ : 10.36, 10.95, 12.89,
13.18, 13.41, 18.39, 20.78 and 23.16 ± 0.2 deg.[Form IV]; or a powder X-ray diffraction pattern having peaks at about 2 θ : 11.08, 12.79,
13.13, 17.29, 19.38, 19.86 and 20.81± 0.2 deg.[Form V]; or a powder X-ray diffraction pattern having peaks at about 2 θ : 10.34, 16.87, 20.34,
20.99, 22.21 and 24.02 ± 0.2 deg. [Form VI]; or a powder X-ray diffraction pattern having peaks at about 2 θ : 5.91, 10.39, 20.53,
20.75, 21.11, 21.60, 27.83 and 28.46 ±0.2 deg. [Form VII]; or a powder X-ray diffraction pattern having peaks at about 2 θ : 5.94, 9.07, 11.79,
13.81, 14.22, 14.99, 15.29 and 15.51 ± 0.2 deg. [Form VIII]
12. A process for preparation of Epinastine HCl Form II comprising the steps of, a) suspending Epinastine in water; b) adding acid to the obtained solution; c) warming the solution to about 65-75 °C; and d) isolating Epinastine HCl Form II.
13. A process for preparation of Epinastine HCl Form III comprising the steps of, a)dissolving Epinastine HCl in a solubilizing solvent selected from the group consisting of alcohols, chlorinated hydrocarbons or aliphatic cyclic ethers; b) adding anti-solvent selected from the group consisting of aliphatic acyclic ethers, hydrocarbons or esters; and c) isolating Epinastine HCl Form III.
14. A process for preparation of Epinastine HCl Form IV comprising the steps of, a) dissolving Epinastine HCl in solubilizing solvent selected from the group consisting of methanol, ethanol or 2-propanol; b) adding anti-solvent selected from the group consisting of diethylether, diisopropylether, methyl tertiary butylether, toluene, xylene, ethyl acetate, butyl acetate or isopropyl acetate to the obtained solution; and c) isolating Epinastine HCl Form IV.
15. A process for preparation of Epinastine HCl Form V comprising the steps of, a) dissolving/suspending Epinastine HCl in a suitable solvent selected from the group consisting of methanol, ethanol, 2-propanol, toluene or xylene; b) adding anti-solvent selected from the group consisting of diethylether, diisopropyl ether or methyl tertiary butyl ether; and d) isolating Epinastine HCl Form V.
16. A process for preparation of Epinastine HCl Form VI comprising the steps of, a) dissolving Epinastine HCl in a solubilizing solvent selected from the group consisting of methanol, ethanol, 2-propanol, 1,4-dioxane, tetrahydrofuran, methylene dichloride or chloroform; b) adding anti-solvent selected from the group consisting of diethylether, diisopropylether, methyl tertiary butylether, pentane, hexane or heptane; and c) isolating Epinastine HCl Form VI.
17. A process for preparation of Epinastine HCl Form VII comprising the steps of, a) dissolving Epinastine HCl in a solubilizing solvent selected from the group consisting of 1,4-dioxane, tetrahydrofuran, dimethyl formamide, dimethyl sulfoxide or N,N-dimethyl acetamide; b) adding anti-solvent selected from the group consisting of acetone, 2-butanone, diethyl ketone, ethyl acetate, butyl acetate or iso propyl acetate; and c) isolating Epinastine HCl Form VII.
18. A process for preparation of Epinastine HCl Form VIII comprising the steps of, a) dissolving Epinastine HCl in a solubilizing solvent selected from the group consisting of chlorinated hydrocarbons or nitriles; b) adding anti-solvent selected from the group consisting aliphatic ketones or ethers; and c) isolating Epinastine HCl Form VIII.
19. Amorphous 3-amino-9,13b-dihydro-lH-dibenz-[c,fJimidazolo[l,5-a]-azepine hydrochloride (Epinastine HCl).
20. A process for preparation of amorphous Epinastine HCl comprising a) dissolving Epinastine HCl in a suitable solvent selected from the group consisting of water, methanol, ethanol, methylene dichloride or chloroform to obtain a solution; and b) spray drying the resultant solution or lyophilizing the resultant solution or evaporating the solvent under vacuum to get amorphous Epinastine HCl.
21. The process as claimed in claim 20 wherein the dissolution is carried out at temperature of 20-40°C, spray drying is carried out at inlet temperature of 40 to 140°C and outlet temperature of 35-85°C, lyophilization is carried out at -20 to - 8O0C and evaporation under vacuum is carried out at 65-850C.
22. A process for preparation of Epinastine HCl Form III comprising, a) suspending Epinastine in first solvent selected from the group consisting of aliphatic alcohols, aliphatic ketones, chlorinated hydrocarbons or aliphatic cyclic ethers; b) adding acid to the obtained mixture; c) pouring the obtained mixture to second solvent selected from aliphatic nitriles, hydrocarbons and esters ; d) isolating Epinastine HCl Form III.
23. A process for preparation of Epinastine HCl Form VIII comprising of, a) suspending Epinastine HCl in first solvent; b) isolating the separated solid; c) resuspending and stirring the separated solid in second solvent; and d) isolating Epinastine HCl Form VIII.
24. The process as claimed in claim 23 wherein said first solvent is acetonitrile and second solvent is selected from ethyl acetate or butyl acetate.
25. Epinastine HCl having d<>o between 25-250μ.
26. Epinastine HCl having dm between 0.5 to 20μ..
27. A process for purification of 3-amino-9,13b-dihydro-lH-dibenzo[c,fJimidazo[l,5- a] azepine hydrobromide (Epinastine hydrobromide) comprising, a) dissolving 3-amino-9,13b-dihydro-lH-dibenzo[c,fJimidazo[l,5-a] azepine hydrobromide in a solvent selected from polar aprotic solvents preferably dimethyl formamide (DMF)5 dimethyl sulfoxide (DMSO), dimethyl acetamide (DMA) at 25-6O0C; b) adding second solvent selected from C1-G* aliphatic ester; and c) isolating pure Epinastine hydrobromide.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IN1872MU2007 | 2007-09-24 | ||
| IN1872/MUM/2007 | 2007-09-24 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2009063504A2 true WO2009063504A2 (en) | 2009-05-22 |
| WO2009063504A3 WO2009063504A3 (en) | 2009-12-03 |
Family
ID=40551912
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IN2008/000612 WO2009063504A2 (en) | 2007-09-24 | 2008-09-24 | Novel crystal modification of epinastine or salts thereof and process for preparation thereof |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2009063504A2 (en) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104098575A (en) * | 2013-04-15 | 2014-10-15 | 四川科瑞德凯华制药有限公司 | Epinastine hydrochloride crystal form, and preparation method and application thereof |
| CN104974164A (en) * | 2015-06-26 | 2015-10-14 | 大连理工大学 | Preparation method of 6-aminomethyl-6,11-dihydro-5H-dibenzo[b,e]azepine |
| CN105153169A (en) * | 2014-11-27 | 2015-12-16 | 兆科药业(广州)有限公司 | Synthesis method for epinastine hydrochloride |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0035749A1 (en) * | 1980-03-08 | 1981-09-16 | C.H. Boehringer Sohn | Heterocyclic compounds, their preparation and pharmaceutical compositions containing them |
-
2008
- 2008-09-24 WO PCT/IN2008/000612 patent/WO2009063504A2/en active Application Filing
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0035749A1 (en) * | 1980-03-08 | 1981-09-16 | C.H. Boehringer Sohn | Heterocyclic compounds, their preparation and pharmaceutical compositions containing them |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104098575A (en) * | 2013-04-15 | 2014-10-15 | 四川科瑞德凯华制药有限公司 | Epinastine hydrochloride crystal form, and preparation method and application thereof |
| CN105153169A (en) * | 2014-11-27 | 2015-12-16 | 兆科药业(广州)有限公司 | Synthesis method for epinastine hydrochloride |
| CN104974164A (en) * | 2015-06-26 | 2015-10-14 | 大连理工大学 | Preparation method of 6-aminomethyl-6,11-dihydro-5H-dibenzo[b,e]azepine |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2009063504A3 (en) | 2009-12-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR101391132B1 (en) | Crystalline minocycline base and processes for its preparation | |
| JP6669932B2 (en) | Vortioxetine pamoate and its crystalline form | |
| WO2021019448A1 (en) | Process for the preparation of transthyretin dissociation inhibitor | |
| WO2018051280A1 (en) | Process for preparation of ribociclib, its acid addition salts | |
| WO2020135058A1 (en) | New crystal form of roxadustat and preparation method thereof | |
| WO2009063504A2 (en) | Novel crystal modification of epinastine or salts thereof and process for preparation thereof | |
| WO2019008520A1 (en) | A process for preparing alectinib or a pharmaceutically acceptable salt thereof | |
| WO2018109786A1 (en) | Novel polymoprphs and salts of polycyclic carbamoyl pyridone derivatives | |
| WO2013024492A2 (en) | A process for the preparation of asenapine and novel salts thereof | |
| EP2688888A1 (en) | Process for the production of a pemetrexed salt | |
| WO2012110947A1 (en) | An improved process for preparation of levonorgestrel | |
| EP3947393B1 (en) | Process for the preparation of midostaurin with high purity | |
| EP2850064B1 (en) | Process for preparation of montelukast sodium | |
| WO2017122139A1 (en) | An improved process for the preparation of pirfenidone | |
| CA2493370C (en) | Crystal forms of olanzapine and processes for their preparation | |
| EP1435359A1 (en) | A process for the purification of roxithromycin | |
| WO2005070939A1 (en) | Synthesis of olanzapine and intermediates thereof | |
| WO2020244148A1 (en) | Doramectin crystal form a, crystal form b, and preparation method thereof | |
| WO2017167949A1 (en) | Crystalline forms of bilastine | |
| JP2002532510A (en) | Macrolide intermediates in the production of clarithromycin | |
| WO2009047796A1 (en) | Polymorphs of imipramine hydrochloride and pamoate salt | |
| JP2002540109A (en) | A novel process for the preparation of crystalline form of doxazosin mesylate designated as Form A | |
| WO2017029584A1 (en) | Amorphous form of bosutinib | |
| KR102522458B1 (en) | Method for Isolation and Purification of Naltrexone | |
| WO2007138376A1 (en) | An improved process for preparing olanzapine form i |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 08849519 Country of ref document: EP Kind code of ref document: A2 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 08849519 Country of ref document: EP Kind code of ref document: A2 |