WO2007072506A2 - Polymorphic forms of dolasetron mesylate and processes thereof - Google Patents
Polymorphic forms of dolasetron mesylate and processes thereof Download PDFInfo
- Publication number
- WO2007072506A2 WO2007072506A2 PCT/IN2006/000499 IN2006000499W WO2007072506A2 WO 2007072506 A2 WO2007072506 A2 WO 2007072506A2 IN 2006000499 W IN2006000499 W IN 2006000499W WO 2007072506 A2 WO2007072506 A2 WO 2007072506A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- dolasetron mesylate
- range
- temperature
- solvent
- dolasetron
- Prior art date
Links
- QTFFGPOXNNGTGZ-RCSCTSIBSA-N u3c8e5bwkr Chemical compound O.CS(O)(=O)=O.C1=CC=C2C(C(OC3C[C@@H]4CC5C[C@@H](N4CC5=O)C3)=O)=CNC2=C1 QTFFGPOXNNGTGZ-RCSCTSIBSA-N 0.000 title claims abstract description 230
- 229960003218 dolasetron mesylate Drugs 0.000 title claims abstract description 162
- 238000000034 method Methods 0.000 title claims abstract description 96
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 claims description 87
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 82
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 claims description 66
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 claims description 51
- 238000000634 powder X-ray diffraction Methods 0.000 claims description 49
- 238000002360 preparation method Methods 0.000 claims description 43
- 239000002904 solvent Substances 0.000 claims description 43
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 40
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 claims description 39
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical group CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 claims description 32
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 claims description 30
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical group CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 claims description 28
- 239000012296 anti-solvent Substances 0.000 claims description 27
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 claims description 24
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 claims description 22
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 claims description 21
- 230000003381 solubilizing effect Effects 0.000 claims description 21
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 claims description 20
- 238000001035 drying Methods 0.000 claims description 20
- 238000001816 cooling Methods 0.000 claims description 16
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 claims description 14
- 239000000203 mixture Substances 0.000 claims description 13
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 claims description 12
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 claims description 12
- 239000003880 polar aprotic solvent Substances 0.000 claims description 11
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 claims description 10
- 239000003586 protic polar solvent Substances 0.000 claims description 9
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical group C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 claims description 8
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 claims description 8
- 239000007921 spray Substances 0.000 claims description 8
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 claims description 7
- 239000000155 melt Substances 0.000 claims description 7
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 claims description 6
- 238000010438 heat treatment Methods 0.000 claims description 6
- 239000000725 suspension Substances 0.000 claims description 6
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 claims description 5
- 125000001931 aliphatic group Chemical group 0.000 claims description 5
- 238000003756 stirring Methods 0.000 claims description 5
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 claims description 4
- 150000001338 aliphatic hydrocarbons Chemical group 0.000 claims description 4
- 150000008280 chlorinated hydrocarbons Chemical group 0.000 claims description 4
- 238000001694 spray drying Methods 0.000 claims description 4
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 claims description 3
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 claims description 3
- 238000001704 evaporation Methods 0.000 claims description 2
- 238000002844 melting Methods 0.000 claims description 2
- 230000008018 melting Effects 0.000 claims description 2
- 238000004519 manufacturing process Methods 0.000 abstract description 7
- 239000000243 solution Substances 0.000 description 83
- 150000001875 compounds Chemical class 0.000 description 53
- 239000007787 solid Substances 0.000 description 53
- 239000011541 reaction mixture Substances 0.000 description 26
- NEDMCWSHHDYQAJ-VGKQMMLZSA-N endo-8-hydroxyhexahydro-1h-2,6-methanoquinolizin-3(2h)-one Chemical compound C1[C@H]2CC(O)C[C@@H]3N2CC(=O)C1C3 NEDMCWSHHDYQAJ-VGKQMMLZSA-N 0.000 description 21
- 229960003413 dolasetron Drugs 0.000 description 20
- CGHRJBLSXVCYQF-YXSUXZIUSA-N dolasetron Chemical compound C1=CC=C[C]2C(C(O[C@@H]3C[C@@H]4C[C@@H]5C[C@@H](N4CC5=O)C3)=O)=CN=C21 CGHRJBLSXVCYQF-YXSUXZIUSA-N 0.000 description 20
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 18
- -1 tetrafluoroborate salt Chemical class 0.000 description 17
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical group [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 15
- KMAKOBLIOCQGJP-UHFFFAOYSA-N indole-3-carboxylic acid Chemical compound C1=CC=C2C(C(=O)O)=CNC2=C1 KMAKOBLIOCQGJP-UHFFFAOYSA-N 0.000 description 14
- 238000010992 reflux Methods 0.000 description 14
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 12
- JCXUVTNCQZYKOG-UHFFFAOYSA-N ethyl 9-(2-ethoxy-2-oxoethyl)-3-oxo-9-azabicyclo[3.3.1]nonane-7-carboxylate Chemical compound C1C(C(=O)OCC)CC2CC(=O)CC1N2CC(=O)OCC JCXUVTNCQZYKOG-UHFFFAOYSA-N 0.000 description 11
- 239000003960 organic solvent Substances 0.000 description 11
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 10
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 10
- 239000000047 product Substances 0.000 description 10
- 150000002118 epoxides Chemical class 0.000 description 9
- CTLAIKSGNQPPLO-UHFFFAOYSA-N ethyl cyclopent-3-ene-1-carboxylate Chemical compound CCOC(=O)C1CC=CC1 CTLAIKSGNQPPLO-UHFFFAOYSA-N 0.000 description 9
- XVSYDLITVYBCBD-UHFFFAOYSA-N cyclopent-3-ene-1-carboxylic acid Chemical compound OC(=O)C1CC=CC1 XVSYDLITVYBCBD-UHFFFAOYSA-N 0.000 description 8
- 238000004090 dissolution Methods 0.000 description 8
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 8
- QAEDZJGFFMLHHQ-UHFFFAOYSA-N trifluoroacetic anhydride Chemical compound FC(F)(F)C(=O)OC(=O)C(F)(F)F QAEDZJGFFMLHHQ-UHFFFAOYSA-N 0.000 description 8
- WCDRGGYHUBTALP-UHFFFAOYSA-N ethyl 9-(2-ethoxy-2-oxoethyl)-3-hydroxy-9-azabicyclo[3.3.1]nonane-7-carboxylate Chemical compound C1C(O)CC2CC(C(=O)OCC)CC1N2CC(=O)OCC WCDRGGYHUBTALP-UHFFFAOYSA-N 0.000 description 7
- 239000000706 filtrate Substances 0.000 description 7
- 229910000029 sodium carbonate Inorganic materials 0.000 description 7
- 235000017550 sodium carbonate Nutrition 0.000 description 7
- 238000006243 chemical reaction Methods 0.000 description 6
- 238000002425 crystallisation Methods 0.000 description 6
- 230000008025 crystallization Effects 0.000 description 6
- 150000002148 esters Chemical class 0.000 description 6
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 6
- 239000012299 nitrogen atmosphere Substances 0.000 description 6
- OXTNCQMOKLOUAM-UHFFFAOYSA-N 3-Oxoglutaric acid Chemical compound OC(=O)CC(=O)CC(O)=O OXTNCQMOKLOUAM-UHFFFAOYSA-N 0.000 description 5
- 239000008186 active pharmaceutical agent Substances 0.000 description 5
- 150000001298 alcohols Chemical class 0.000 description 5
- 239000007864 aqueous solution Substances 0.000 description 5
- DJQLJFSLFWCBAU-UHFFFAOYSA-N ethyl 2-(3-hydroxy-9-azabicyclo[3.3.1]nonan-5-yl)acetate Chemical compound C1CCC2CC(O)CC1(CC(=O)OCC)N2 DJQLJFSLFWCBAU-UHFFFAOYSA-N 0.000 description 5
- 238000004108 freeze drying Methods 0.000 description 5
- FJBKMTQUKYFZDN-UHFFFAOYSA-N methyl 4-oxo-2-(2-oxoethyl)butanoate Chemical compound COC(=O)C(CC=O)CC=O FJBKMTQUKYFZDN-UHFFFAOYSA-N 0.000 description 5
- 150000007524 organic acids Chemical class 0.000 description 5
- 229910000027 potassium carbonate Inorganic materials 0.000 description 5
- 235000011181 potassium carbonates Nutrition 0.000 description 5
- IWZKICVEHNUQTL-UHFFFAOYSA-M potassium hydrogen phthalate Chemical compound [K+].OC(=O)C1=CC=CC=C1C([O-])=O IWZKICVEHNUQTL-UHFFFAOYSA-M 0.000 description 5
- 238000000746 purification Methods 0.000 description 5
- 239000012279 sodium borohydride Substances 0.000 description 5
- 229910000033 sodium borohydride Inorganic materials 0.000 description 5
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 description 4
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 4
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 4
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 4
- 150000004292 cyclic ethers Chemical group 0.000 description 4
- 238000006114 decarboxylation reaction Methods 0.000 description 4
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 4
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 4
- 125000001181 organosilyl group Chemical group [SiH3]* 0.000 description 4
- KHIWWQKSHDUIBK-UHFFFAOYSA-N periodic acid Chemical compound OI(=O)(=O)=O KHIWWQKSHDUIBK-UHFFFAOYSA-N 0.000 description 4
- 150000008302 pseudopelletierine derivatives Chemical class 0.000 description 4
- HUHXLHLWASNVDB-UHFFFAOYSA-N 2-(oxan-2-yloxy)oxane Chemical compound O1CCCCC1OC1OCCCC1 HUHXLHLWASNVDB-UHFFFAOYSA-N 0.000 description 3
- BUDQDWGNQVEFAC-UHFFFAOYSA-N Dihydropyran Chemical compound C1COC=CC1 BUDQDWGNQVEFAC-UHFFFAOYSA-N 0.000 description 3
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- 150000002009 diols Chemical class 0.000 description 3
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 3
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- 238000007738 vacuum evaporation Methods 0.000 description 3
- TXTWXQXDMWILOF-UHFFFAOYSA-N (2-ethoxy-2-oxoethyl)azanium;chloride Chemical compound [Cl-].CCOC(=O)C[NH3+] TXTWXQXDMWILOF-UHFFFAOYSA-N 0.000 description 2
- 238000005160 1H NMR spectroscopy Methods 0.000 description 2
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 2
- 239000004471 Glycine Substances 0.000 description 2
- IYTXKIXETAELAV-UHFFFAOYSA-N Nonan-3-one Chemical compound CCCCCCC(=O)CC IYTXKIXETAELAV-UHFFFAOYSA-N 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical group CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 2
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 2
- KXKVLQRXCPHEJC-UHFFFAOYSA-N acetic acid trimethyl ester Natural products COC(C)=O KXKVLQRXCPHEJC-UHFFFAOYSA-N 0.000 description 2
- 238000005903 acid hydrolysis reaction Methods 0.000 description 2
- 239000011260 aqueous acid Substances 0.000 description 2
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 2
- 238000009833 condensation Methods 0.000 description 2
- 230000005494 condensation Effects 0.000 description 2
- 239000013078 crystal Substances 0.000 description 2
- NKDDWNXOKDWJAK-UHFFFAOYSA-N dimethoxymethane Chemical compound COCOC NKDDWNXOKDWJAK-UHFFFAOYSA-N 0.000 description 2
- 150000002170 ethers Chemical class 0.000 description 2
- 125000003754 ethoxycarbonyl group Chemical group C(=O)(OCC)* 0.000 description 2
- ZYXAXBVALNQLAN-UHFFFAOYSA-N ethyl 4-oxo-2-(2-oxoethyl)butanoate Chemical compound CCOC(=O)C(CC=O)CC=O ZYXAXBVALNQLAN-UHFFFAOYSA-N 0.000 description 2
- 238000001914 filtration Methods 0.000 description 2
- 235000019253 formic acid Nutrition 0.000 description 2
- 239000007789 gas Substances 0.000 description 2
- 238000004128 high performance liquid chromatography Methods 0.000 description 2
- 229910010272 inorganic material Inorganic materials 0.000 description 2
- 239000011147 inorganic material Substances 0.000 description 2
- 239000000543 intermediate Substances 0.000 description 2
- JMMWKPVZQRWMSS-UHFFFAOYSA-N isopropanol acetate Natural products CC(C)OC(C)=O JMMWKPVZQRWMSS-UHFFFAOYSA-N 0.000 description 2
- 229940011051 isopropyl acetate Drugs 0.000 description 2
- GWYFCOCPABKNJV-UHFFFAOYSA-M isovalerate Chemical compound CC(C)CC([O-])=O GWYFCOCPABKNJV-UHFFFAOYSA-M 0.000 description 2
- 125000000468 ketone group Chemical group 0.000 description 2
- SIAPCJWMELPYOE-UHFFFAOYSA-N lithium hydride Chemical compound [LiH] SIAPCJWMELPYOE-UHFFFAOYSA-N 0.000 description 2
- 229910000103 lithium hydride Inorganic materials 0.000 description 2
- PSHKMPUSSFXUIA-UHFFFAOYSA-N n,n-dimethylpyridin-2-amine Chemical compound CN(C)C1=CC=CC=N1 PSHKMPUSSFXUIA-UHFFFAOYSA-N 0.000 description 2
- 229910000489 osmium tetroxide Inorganic materials 0.000 description 2
- 239000012285 osmium tetroxide Substances 0.000 description 2
- FDPIMTJIUBPUKL-UHFFFAOYSA-N pentan-3-one Chemical compound CCC(=O)CC FDPIMTJIUBPUKL-UHFFFAOYSA-N 0.000 description 2
- 239000000825 pharmaceutical preparation Substances 0.000 description 2
- 229940127557 pharmaceutical product Drugs 0.000 description 2
- TYJJADVDDVDEDZ-UHFFFAOYSA-M potassium hydrogencarbonate Chemical compound [K+].OC([O-])=O TYJJADVDDVDEDZ-UHFFFAOYSA-M 0.000 description 2
- 229920006395 saturated elastomer Polymers 0.000 description 2
- 229910001494 silver tetrafluoroborate Inorganic materials 0.000 description 2
- JQWHASGSAFIOCM-UHFFFAOYSA-M sodium periodate Chemical compound [Na+].[O-]I(=O)(=O)=O JQWHASGSAFIOCM-UHFFFAOYSA-M 0.000 description 2
- 238000010626 work up procedure Methods 0.000 description 2
- DNIAPMSPPWPWGF-GSVOUGTGSA-N (R)-(-)-Propylene glycol Chemical compound C[C@@H](O)CO DNIAPMSPPWPWGF-GSVOUGTGSA-N 0.000 description 1
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 1
- IIJFYTVJRDKVCI-UHFFFAOYSA-N 1h-indole-3-carbonyl chloride Chemical compound C1=CC=C2C(C(=O)Cl)=CNC2=C1 IIJFYTVJRDKVCI-UHFFFAOYSA-N 0.000 description 1
- MEKOFIRRDATTAG-UHFFFAOYSA-N 2,2,5,8-tetramethyl-3,4-dihydrochromen-6-ol Chemical compound C1CC(C)(C)OC2=C1C(C)=C(O)C=C2C MEKOFIRRDATTAG-UHFFFAOYSA-N 0.000 description 1
- LTMRRSWNXVJMBA-UHFFFAOYSA-L 2,2-diethylpropanedioate Chemical compound CCC(CC)(C([O-])=O)C([O-])=O LTMRRSWNXVJMBA-UHFFFAOYSA-L 0.000 description 1
- XVMSFILGAMDHEY-UHFFFAOYSA-N 6-(4-aminophenyl)sulfonylpyridin-3-amine Chemical compound C1=CC(N)=CC=C1S(=O)(=O)C1=CC=C(N)C=N1 XVMSFILGAMDHEY-UHFFFAOYSA-N 0.000 description 1
- DKPFZGUDAPQIHT-UHFFFAOYSA-N Butyl acetate Natural products CCCCOC(C)=O DKPFZGUDAPQIHT-UHFFFAOYSA-N 0.000 description 1
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- 238000005033 Fourier transform infrared spectroscopy Methods 0.000 description 1
- LFTLOKWAGJYHHR-UHFFFAOYSA-N N-methylmorpholine N-oxide Chemical compound CN1(=O)CCOCC1 LFTLOKWAGJYHHR-UHFFFAOYSA-N 0.000 description 1
- 206010028980 Neoplasm Diseases 0.000 description 1
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 1
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 1
- 206010047700 Vomiting Diseases 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 230000001476 alcoholic effect Effects 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 239000002111 antiemetic agent Substances 0.000 description 1
- 229940125683 antiemetic agent Drugs 0.000 description 1
- 239000002579 antinauseant Substances 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- MIOPJNTWMNEORI-UHFFFAOYSA-N camphorsulfonic acid Chemical compound C1CC2(CS(O)(=O)=O)C(=O)CC1C2(C)C MIOPJNTWMNEORI-UHFFFAOYSA-N 0.000 description 1
- 201000011510 cancer Diseases 0.000 description 1
- 235000011089 carbon dioxide Nutrition 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 239000003610 charcoal Substances 0.000 description 1
- 238000002512 chemotherapy Methods 0.000 description 1
- 238000004440 column chromatography Methods 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- RXKJFZQQPQGTFL-UHFFFAOYSA-N dihydroxyacetone Chemical compound OCC(=O)CO RXKJFZQQPQGTFL-UHFFFAOYSA-N 0.000 description 1
- BEPAFCGSDWSTEL-UHFFFAOYSA-N dimethyl malonate Chemical compound COC(=O)CC(=O)OC BEPAFCGSDWSTEL-UHFFFAOYSA-N 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 229940088679 drug related substance Drugs 0.000 description 1
- 230000000095 emetic effect Effects 0.000 description 1
- NTNZTEQNFHNYBC-UHFFFAOYSA-N ethyl 2-aminoacetate Chemical compound CCOC(=O)CN NTNZTEQNFHNYBC-UHFFFAOYSA-N 0.000 description 1
- NHCULWYCYWXTRR-UHFFFAOYSA-N ethyl 7-tert-butyl-1-dimethylsilyloxy-9-(2-ethoxy-2-oxoethyl)-7-hydroxy-9-azabicyclo[3.3.1]nonane-3-carboxylate Chemical compound C1C(C(=O)OCC)CC2(O[SiH](C)C)CC(O)(C(C)(C)C)CC1N2CC(=O)OCC NHCULWYCYWXTRR-UHFFFAOYSA-N 0.000 description 1
- 238000003810 ethyl acetate extraction Methods 0.000 description 1
- OAYLNYINCPYISS-UHFFFAOYSA-N ethyl acetate;hexane Chemical compound CCCCCC.CCOC(C)=O OAYLNYINCPYISS-UHFFFAOYSA-N 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 238000004817 gas chromatography Methods 0.000 description 1
- FUZZWVXGSFPDMH-UHFFFAOYSA-M hexanoate Chemical compound CCCCCC([O-])=O FUZZWVXGSFPDMH-UHFFFAOYSA-M 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 229910017053 inorganic salt Inorganic materials 0.000 description 1
- 238000011835 investigation Methods 0.000 description 1
- 150000002576 ketones Chemical group 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- LULAYUGMBFYYEX-UHFFFAOYSA-N metachloroperbenzoic acid Natural products OC(=O)C1=CC=CC(Cl)=C1 LULAYUGMBFYYEX-UHFFFAOYSA-N 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- OIXMUQLVDNPHNS-UHFFFAOYSA-N methanesulfonic acid;hydrate Chemical compound O.CS(O)(=O)=O OIXMUQLVDNPHNS-UHFFFAOYSA-N 0.000 description 1
- ODOKMFOOOYEECW-UHFFFAOYSA-N methyl 3-hydroxy-3-(methoxymethoxy)-9-(2-methoxy-2-oxoethyl)-9-azabicyclo[3.3.1]nonane-7-carboxylate Chemical compound C1C(C(=O)OC)CC2CC(OCOC)(O)CC1N2CC(=O)OC ODOKMFOOOYEECW-UHFFFAOYSA-N 0.000 description 1
- YOHHRDUPBZSBFP-UHFFFAOYSA-N methyl 9-(2-methoxy-2-oxoethyl)-3-oxo-9-azabicyclo[3.3.1]nonane-7-carboxylate Chemical compound C1C(C(=O)OC)CC2CC(=O)CC1N2CC(=O)OC YOHHRDUPBZSBFP-UHFFFAOYSA-N 0.000 description 1
- CEOILRYKIJRPBZ-UHFFFAOYSA-N methyl cyclopent-3-ene-1-carboxylate Chemical compound COC(=O)C1CC=CC1 CEOILRYKIJRPBZ-UHFFFAOYSA-N 0.000 description 1
- MCSAJNNLRCFZED-UHFFFAOYSA-N nitroethane Chemical compound CC[N+]([O-])=O MCSAJNNLRCFZED-UHFFFAOYSA-N 0.000 description 1
- 150000002894 organic compounds Chemical class 0.000 description 1
- 238000005949 ozonolysis reaction Methods 0.000 description 1
- 239000011736 potassium bicarbonate Substances 0.000 description 1
- 235000015497 potassium bicarbonate Nutrition 0.000 description 1
- 229910000028 potassium bicarbonate Inorganic materials 0.000 description 1
- 235000011118 potassium hydroxide Nutrition 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 230000005855 radiation Effects 0.000 description 1
- 230000008929 regeneration Effects 0.000 description 1
- 238000011069 regeneration method Methods 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 239000003369 serotonin 5-HT3 receptor antagonist Substances 0.000 description 1
- 229940100890 silver compound Drugs 0.000 description 1
- 150000003379 silver compounds Chemical class 0.000 description 1
- 125000003808 silyl group Chemical group [H][Si]([H])([H])[*] 0.000 description 1
- 239000002002 slurry Substances 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- HRZFUMHJMZEROT-UHFFFAOYSA-L sodium disulfite Chemical compound [Na+].[Na+].[O-]S(=O)S([O-])(=O)=O HRZFUMHJMZEROT-UHFFFAOYSA-L 0.000 description 1
- 235000011121 sodium hydroxide Nutrition 0.000 description 1
- 239000004296 sodium metabisulphite Substances 0.000 description 1
- 235000010262 sodium metabisulphite Nutrition 0.000 description 1
- 229910052938 sodium sulfate Inorganic materials 0.000 description 1
- 235000011152 sodium sulphate Nutrition 0.000 description 1
- 210000002784 stomach Anatomy 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- ILMRJRBKQSSXGY-UHFFFAOYSA-N tert-butyl(dimethyl)silicon Chemical group C[Si](C)C(C)(C)C ILMRJRBKQSSXGY-UHFFFAOYSA-N 0.000 description 1
- BCNZYOJHNLTNEZ-UHFFFAOYSA-N tert-butyldimethylsilyl chloride Chemical compound CC(C)(C)[Si](C)(C)Cl BCNZYOJHNLTNEZ-UHFFFAOYSA-N 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 125000000025 triisopropylsilyl group Chemical group C(C)(C)[Si](C(C)C)(C(C)C)* 0.000 description 1
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/12—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains three hetero rings
- C07D471/18—Bridged systems
Definitions
- the present disclosure relates to novel crystalline polymorphs of Dolasetron mesylate having formula (1) and industrial processes for producing the same. Further, it discloses processes for producing Form I of Dolasetron mesylate. Furthermore, the present disclosure teaches novel amorphous form and industrial processes for producing amorphous form.
- Dolasetron mesylate is an antinauseant and antiemetic agent. It is a selective serotonin 5-HT 3 receptor antagonist and is indicated for the prevention of nausea and vomiting associated with emetogenic cancer chemotherapy.
- EP0266730/US4906755 describes process for the preparation endo-hexahydro-8-(3- indolylcarbonyloxy)-2,6-methano-2H-qumolizin-3(4H)-one methanesulfonate or Dolasetron mesylate (1) by the condensation of diethyl malonate with cis-l,4-dichloro-2- butene (2) in presence of lithium hydride in dimethylformamide to give diethyl-3- cyclopentene-l,l-dicarboxylate (3), which on hydrolysis and decarboxylation gave 3- cyclopentene-1-carboxylic acid (4).
- the compound (4) was further treated with thionyl chloride and pyridine in ethanol to obtain ethyl 3-cyclopentene-l-carboxylate (5).
- Compound (5) was oxidized to 4-ethoxycarbonyl-l, 2-cyclopentanediol (6) by using N- methylmorpholine N-oxide in the presence of osmium tetroxide catalyst.
- the diol (6) was cleaved to the /3-ethoxycarbonylglutaraldehyde (7) using sodium periodate and used, directly in the next reaction.
- the reduced alcohol (9) was treated with dihydropyran to protect the hydroxyl group as a tetrahydropyranyl ether (10). Dieckmann cyclisation of the compound (10) using strong base (potassium t- butoxide) followed by aqueous acid hydrolysis and decarboxylation gave the desired alcohol.
- the resulting alcohols can exist in two conformations - axial and equatorial.
- the main product obtained by above procedure was the axial alcohol or endo-hexahydro-8- hydroxy-2,6-methano-2H-quinolizin-3-(4H)-one (11) and it can be separated from the equatorial isomer by crystallization of the camphorsulfonate or tetrafluoroborate salt.
- the tetrafluoroborate salt of endo-hexahydro-8-hydroxy-2,6-methano-2H-quinolizin-3-(4H)- one (11) was further reacted with 3-indolecarboxylic acid chloride in presence of silver tetrafluoroborate in anhydrous nitroethane at -78 0 C to endo-hexahydro-8-(3- indolylcarbonyloxy)-2,6-methano-2H-quinolizin-3(4H)-one or Dolasetron base, which was further converted into Dolasetron mesylate monohydrate (Scheme I) with a yield of 66%. No further purification is described.
- the above process uses column chromatography for purification of compounds (9) and (10), which is expensive, time consuming and impractical on an industrial scale.
- the above patent does not disclose the yield and purity of Dolasetron mesylate obtained and so also for the intermediates.
- Osmium tetroxide used for preparation of compound (6) is toxic, has a corrosive action on eyes and hence difficult to use at industrial scale.
- this process uses high volume of water during preparation of the compound (8); preparation of compound (11) from compound (10) is tedious, because the workup involves several extractions with ethyl acetate and preparation of compound (1) in presence of silver tetrafluoroborate involves the use of expensive silver compound.
- EP0339669 provides a process for the preparation of endo-hexahydro-8-(3-indolylcarbonyloxy)-2,6-methano-2H-quinolizin-3(4H)-one methanesulfonate or Dolasetron mesylate (1) by the condensation of dimethyl malonate with cis-l,4-dichloro-2-butene (2) in presence of lithium hydride in dimethyl formamide to give dimethyl-3-cyclopentene-l,l-dicarboxylate (12), which was decarboxymethylated to obtain methyl-3-cyclopentene-l-carboxylate (13).
- This compound (13) was treated with m-chloroperbenzoic acid in dichloromethane to obtain l-methoxycarbonyl-3- cyclopenteneoxide (14).
- the compound (13) on ozonolysis gave ⁇ - methoxycarbonylglutaraldehyde (15) or the epoxide (14) was reacted with periodic acid to obtain the ⁇ -methoxycarbonylglutaraldehyde (15), which was used directly in the next reaction.
- Robinson-Schopf cyclisation of the compound (15) with potassium hydrogen phthalate, acetonedicarboxylic acid and glycine ethyl ester hydrochloride gave the pseudopelletierine derivative i.e.
- the reduced alcohol (17) was treated with dihydropyran to protect the hydroxyl group as a tetrahydropyranyl ether (18a) or treated with methylal to protect the hydroxyl group to obtain 3-methoxymethoxy- 7-methoxycarbonyl-9-(methoxycarbonylmethyl)-9-azabicyclo[3.3.1]nonan-3-ol (18b).
- the alcohol (11) was further reacted with 3-indolecarboxylic acid in presence of trifluoroacetic anhydride in dichloromethane to endo-hexahydro-8-(3-indolylcarbonyloxy)-2, 6-methano-2H- quinolizin-3(4H)-one or Dolasetron base, which was then converted into Dolasetron mesylate (1) (Scheme II) by treating with methanesulphonicacid in acetone. Further, crude Dolasetron mesylate (1) was dissolved in aqueous isopropanol and regenerated by adding ether to obtain Dolasetron mesylate (1) with a yield of 85.90 %. Disadvantages of this process are:
- EP 0266730 involves treatment of endo-hexahydro-8- (3-indolylcarbonyloxy)-2, 6-methano-2H-quinolizin-3(4H)-one with a solution of methane sulfonic acid in ethanol to provide Dolasetron mesylate monohydrate.
- EP 0339669 describes crystallization of crude Dolasetron mesylate by dissolution in aqueous isopropanol and regeneration by adding ether.
- the polymorphic form obtained by the processes described in US 4906755/EP 0266730 and EP 0339669 is designated herein as Dolasetron mesylate Form I.
- XRPD of Dolasetron mesylate Form I is disclosed in Figure 1.
- the ability of the compound to exhibit more than one orientation or conformation of molecule within the crystal lattice is called polymorphism.
- Many organic compounds including active pharmaceutical ingredients (API's) exhibit polymorphism.
- Drug substance existing in various polymorphic forms differs from each other in terms of stability, solubility, compressibility, flowability and spectroscopic properties, thus affecting dissolution, bioavailability and handling characteristics of the substance.
- Rate of dissolution of an API's in patient's stomach fluid can have therapeutic consequences since it imposes an upper limit on the rate at which an orally administrated API can reach the patient . bloodstream. Flowability affects the ease with which the material is handled while processing a pharmaceutical product.
- XRPD X-Ray powder diffraction
- FT- IR Fourier transformer Infrared
- Solid State 13 C-NMR Solid State 13 C-NMR
- Another object is to provide a process for preparation of Dolasetron mesylate polymorphic Form I. It is also an object of to provide novel amorphous form of Dolasetron mesylate and industrial processes for producing it.
- the present disclosure provides a process for the preparation of a crystalline polymorphic Form I of endo-hexahydro-8-(3-indolylcarbonyloxy)-2, 6- methano-2H-quinolizin-3 (4H)-one methanesulfonate (Dolasetron rrfesy ⁇ ate).
- the present invention provides a crystalline polymorphic Form II of endo-hexahydro-8-(3-indolylcarbonyloxy)-2,6-methano-2H-quinolizm-3(4H)-one methanesulfonate, Dolasetron mesylate.
- the present invention provides a process for producing, polymorphic Form II of Dolasetron mesylate.
- the present invention provides a crystalline polymorphic Form III of endo-hexahydro-8-(3-indolylcarbonyloxy)-2,6-methano-2H-quinolizin-3 (4H)-one methanesulfonate, Dolasetron mesylate.
- the present invention provides a process for producing polymorphic Form III of Dolasetron mesylate.
- the present invention provides a crystalline polymorphic Form IV of endo-hexahydro-8-(3-indolylcarbonyloxy)-2,6-methano-2H-quinolizin-3(4H)-one methanesulfonate, Dolasetron mesylate.
- the present invention relates to a process for producing polymorphic Form IV of Dolasetron mesylate.
- the present invention provides a crystalline polymorphic Form V of endo-hexahydro ⁇ 8-(3 -indolylcarbonyloxy)-2,6-methano-2H-quinolizin-3 (4H)-one methanesulfonate, Dolasetron mesylate.
- the present invention provides a process for producing polymorphic Form V of Dolasetron mesylate.
- the present invention provides a crystalline polymorphic Form VI of endo-hexahydro-8-(3-indolylcarbonyloxy)-2,6-methano-2H-quinolizin- 3(4H)-one methanesulfonate, Dolasetron mesylate.
- the present invention provides a process for producing polymorphic Form VI of Dolasetron mesylate.
- the present invention provides a crystalline polymorphic Form VII of endo-hexahydro-8-(3-indolylcarbonyloxy)-2,6-methan ⁇ -2H-quinolizin-3(4H)-one methanesulfonate, Dolasetron mesylate.
- the present invention provides a process for producing polymorphic Form VII of Dolasetron mesylate.
- the present invention provides a crystalline polymorphic Form VIII of endo-hexahydro-8-(3-indolylcarbonyloxy)-2,6-methano-2H-quinolizm- 3(4H)-one methanesulfonate, Dolasetron mesylate.
- the present invention provides a process for producing polymorphic Form VIII of Dolasetron mesylate.
- the present invention provides a crystalline polymorphic Form IX of endo-hexahydro-8-(3-indolylcarbonyloxy)-2,6-methano-2H-quinolizin-3(4H)-one methanesulfonate, Dolasetron mesylate.
- the present invention provides a process for producing polymorphic Form K of Dolasetron mesylate.
- the present invention provides an amorphous form of endo- hexahydro-8-(3-indolylcarbonyloxy)-2, 6-methano-2H-quinolizin-3 (4H)-one methanesulfonate, Dolasetron mesylate.
- the present invention provides processes for preparation of amorphous form of Dolasetron mesylate.
- Figure 1 shows XRPD pattern of Dolasetron mesylate Form I
- Figure 2 shows XRPD pattern of Dolasetron mesylate Form II
- Figure 3 shows XRPD pattern of Dolasetron mesylate Form III
- Figure 4 shows XRPD pattern of Dolasetron mesylate Form IV
- FIG. 5 shows XRPD pattern of Dolasetron mesylate Form V
- Figure 6 shows XRPD pattern of Dolasetron mesylate Form VI
- Figure 7 shows XRPD pattern of Dolasetron mesylate Form VII
- Figure 8 shows XRPD pattern of Dolasetron mesylate Form VIII
- Figure 9 shows XRPD pattern of Dolasetron mesylate Form IX
- Figure 10 shows XRPD pattern of amorphous form of Dolasetron mesylate DETAILED DESCRIPTION OF THE INVENTION
- the present disclosure relates to a process for preparation of Dolasetron mesylate or endo-hexahydro-8-(3-indolylcarbonyloxy)-2, 6-methano-2H-quinolizin-3 (4H)-one methanesulfonate of the formula (1) in high yield and high purity comprising:
- Formula (V) b. treating the compound haying the structural formula (V) with m-chloroperbenzoic acid in dichloromethane to give an epoxide having the structural formula (XIX),
- Formula (VII) d cyclising the compound having the structural formula (VII) with potassium hydrogen phthalate, acetonedicarboxylic acid and glycine ester hydrochloride by Robinson- Schopf cyclisation to obtain pseudopelletierine derivative having the structural formula (VIII);
- Formula (VIII) e. reducing the compound having the structural formula (VIII) with sodiumborohydride in alcohol followed by treatment with an organic acid to obtain compound having the structural formula (IX);
- Formula (IX) f. protecting the compound having the structural formula (IX) as a silyl derivative having the structural formula (XX) by treating it with a silyl halide in an organic solvent (wherein Z silyl group);
- Scheme III depicts a process for the preparation of 3-cyclopentene-l-carboxylic acid ester (5) is disclosed, said process comprising: reacting 3-cyclopentene-l-carboxylic acid (4) with anhydrous HCl gas or concentrated hydrochloric acid or thionyl chloride in an alcohol, wherein the alcohol is either methanol or ethanol; treating the compound (5) with m-chloroperbenzoic acid in a solvent selected from dichloromethane and ethyl acetate to obtain the corresponding epoxide (19); reacting the compound (19) with periodic acid under nitrogen atmosphere to obtain compound (7); treating the compound (7) with potassium hydrogen phthalate, acetonedicarboxylic acid and glycine ester hydrochloride in water to obtain pseudopelletierine derivative (8); reducing the compound (8) with sodiumborohydride in an alcohol and further treating with an organic acid to obtain compound (9), wherein the organic acid is selected from formic acid, methane
- a major advantage of the use of silyl protecting group is that it yields greater than 95 % of compound (20) as compared to, use of dihydropyran (75%) or methylal (84%).
- the compound (20) is treated with a strong base in toluene and further treated with an organic acid in an organic solvent to form compound (21).
- the organic solvent is selected from halogenated solvents, ethers and esters.
- the organic solvent is preferably selected from methylene chloride, chloroform, ethyl acetate, isopropyl acetate, diethyl ether, diisopropyl ether or mixtures thereof.
- the organic acid is selected from formic acid and acetic acid.
- the compound (21) is heated with hydrochloric acid in water to give compound (11). Hydrochloric acid and water are used in the ratio of 1:2 volumes. The ratio of compound (21) to water in the reaction is about 1: 8 to 1:10.
- the reaction mixture is concentrated and the residue obtained is treated with an organic solvent and filtered. The filtrate is concentrated to obtain compound (11).
- the organic solvent is selected from alcohols and halogenated solvent preferably methanol, ethanol, isopropanol, n-butanol, dichloromethane, chloroform or mixture thereof.
- the reaction mixture is extracted with an organic solvent selected from ethyl acetate, isopropanol or n-butanol. Alternately the reaction mixture is saturated with an inorganic salt and extracted with an organic solvent selected from ethyl acetate or n-butanol or isopropanol.
- Dolasetron base is reacted with indole-3-carboxylic acid in presence of trifiuoroacetic acid anhydride in dichloromethane to give Dolasetron base.
- the ratio of indole-3-carboxylic acid and trifluoro acetic anhydride used is in the range of 1:1.1 to 1:2.
- Dolasetron base thus obtained is isolated by conventional method.
- Dolasetron base is solubilized in acetone and converted into its mesylate salt using methane sulphonic acid.
- the resultant mesylate salt is dissolved in water and extracted with a halogenated solvent or ester to remove traces of impurity.
- the halogenated solvent is selected from dichloromethane and chloroform, and the ester is selected from methyl acetate, ethyl acetate and isopropyl acetate.
- the aqueous layer is basified with a base to obtain Dolasetron base.
- the base is selected from sodium carbonate, potassium carbonate, sodium bicarbonate, potassium bicarbonate, sodium hydroxide, potassium hydroxide or mixtures thereof.
- Dolasetron base thus obtained is treated with methane sulphonic acid in a mixture of acetone and water to provide Dolasetron Mesylate. Polymorphic forms of Dolasetron mesylate
- One more embodiment of the invention provides novel crystalline polymorphic forms, viz: Form II, Form III, Form IV, Form V, Form VI, Form VII, Form VIII and Form IX. Another embodiment of the invention provides a process for manufacturing these crystalline polymorphic forms.
- the invention also discloses novel amorphous form of Dolasetron mesylate.
- the polymorphic Form I of Dolasetron mesylate is obtained by crystallization. The process involves, dissolving Dolasetron mesylate in a solvent selected from aliphatic alcohols, aliphatic ketones, aliphatic esters, aliphatic nitriles or mixtures thereof at a temperature in the range of 30°C-80°C to yield a clear solution.
- the clear solution is cooled at a temperature in the range of 0°C-30°C, preferably in the range of 25°C-30°C to i obtain a solid.
- Dolasetron mesylate form I is obtained by filtering and drying the solid at a temperature in the range of 30°C-90°C, preferably in the range of 60°C-70°C.
- the aliphatic alcohol selected is isopropanol; the aliphatic ester is selected from methyl acetate, ethyl acetate and butyl acetate; aliphatic ketone is selected from acetone, 2- butanone, diethylketone, and the like; and aliphatic nitrile is acetonitrile.
- Dolasetron mesylate Form I is also obtained by solvent and anti-solvent process.
- the said process comprises: dissolving Dolasetron mesylate in a solubilizing solvent, adding an anti-solvent, stirring the suspension with or without cooling, isolating and drying the product at 50°C-70°C.
- the solubilizing solvents selected for dissolution are polar aprotic solvents.
- the polar aprotic solvent is selected from N, N-dimethyl formamide, dimethyl sulfoxide and N, N-dimethyl acetamide.
- the anti-solvent is selected from cyclic ethers, aromatic hydrocarbons and alcohols.
- the cyclic ether selected is tetrahydrofuran.
- the aromatic hydrocarbon is toluene and the alcohol is isopropanol.
- the polymorphic Form II is obtained by crystallizing Dolasetron mesylate from methanol.
- the polymorphic Form II is obtained by dissolving Dolasetron mesylate in methanol at a temperature in the range of 30-80 0 C, preferably in the range of 60-70 0 C, cooling the solution at a temperature in the range of -5°C to 25 0 C, preferably in the range of 2 to 7 0 C, isolating and drying the product at a temperature in the range of in the range of 40-90 0 C, preferably in the range of in the range of 60-70 0 C.
- the XRPD of Dolasetron mesylate Form II is given in Figure 2.
- the XRPD of Dolasetron mesylate Form II exhibits following peaks:
- the polymorphic Form III is obtained by crystallizing Dolasetron mesylate from ethanol.
- the polymorphic Form III is obtained by dissolving Dolasetron mesylate in ethanol at a temperature in the range of 30-80 0 C, preferably in the range of 75-8O 0 C, cooling the solution at a temperature in the range of-5°C to 25°C, preferably in the range of 2 to 7°C, isolating and drying the product at a temperature in the range of in the range of 40-90 0 C, preferably in the range of 60-70 0 C.
- the polymorphic Form III is obtained by using solvent and anti-solvent combination process.
- the said process comprises: dissolving Dolasetron mesylate in a solubilizing solvent at a temperature in the range of 30-80 0 C, preferably in the range of 60-80 0 C, adding an anti-solvent at a temperature in the range of 30-55 0 C, preferably in the range of 40-50 0 C, isolating and drying the product at a temperature in the range of 40- 90 0 C, preferably in the range of 60-70 0 C.
- the solubilizing solvents are selected from lower aliphatic alcohols.
- the lower alcohol is selected from methanol, ethanol, n-propanol and isopropanol, preferably ethanol.
- the anti-solvent is selected from aliphatic hydrocarbons n-pentane, n-hexane and n-heptane, preferably n-hexane.
- the XRPD of Dolasetron mesylate Form III is given in Figure 3.
- the XRPD of Dolasetron mesylate Form III exhibits following peaks:
- Polymorphic Form IV is obtained by crystallizing Dolasetron mesylate from n- propanol.
- the polymorphic Form IV is obtained by dissolving Dolasetron mesylate in n- propanol at a temperature in the range of 30-100 0 C, preferably in the range of 90-100 0 C, cooling the solution at a temperature in the range of -5°C to 25 0 C, preferably in the range of 2 to 7°C, isolating and drying the product at a temperature in the range of in the range of 40-90 0 C, preferably in the range of in the range of 60-70 0 C.
- the XRPD of Dolasetron mesylate Form IV is given in Figure 4.
- the XRPD of Dolasetron mesylate Form IV exhibits following peaks:
- Polymorphic Form V is obtained by crystallizing Dolasetron mesylate from chlorinated hydrocarbons.
- the polymorphic Form V is obtained by dissolving Dolasetron mesylate in a solubilizing solvent at a temperature in the range of 30-80 0 C, cooling the solution at a temperature in the range of-5°C to 30 0 C, preferably in the range of 25-3O 0 C, isolating and drying the product at a temperature in the range of in the range of 40-90 0 C, preferably in the range of in the range of 60-70 0 C.
- the solubilizing solvent is chlorinated hydrocarbon and is selected from methylene dichloride or chloroform.
- Dolasetron mesylate Form V is given in Figure 5.
- the XRPD of Dolasetron mesylate Form V exhibits following peaks:
- Polymorphic Form VI is obtained from Dolasetron mesylate by solvent and anti- solvent combination process.
- the said process comprises of dissolving Dolasetron mesylate in a solubilizing solvent like a polar aprotic solvent at a temperature in the range of 20-35 0 C, preferably in the range of 25-30 0 C, adding an anti-solvent at a temperature in the range of 20-45 0 C, preferably in the range of 25-30 0 C, isolating and drying the product at a temperature in the range of 40-90 0 C, preferably in the range of 60-70 0 C.
- the polar aprotic solvent selected for dissolving Dolasetron mesylate is dimethyl formamide or dimethyl sulfoxide.
- the anti-solvent is selected from cyclic ethers such as 1, 4-dioxane.
- the XRPD of Dolasetron mesylate Form VI is given in Figure 6.
- the XRPD of Dolasetron mesylate Form VI exhibits following peaks: Position [°2 ⁇ ] ReI. Int. [%]
- the polymorphic Fo ⁇ n VII is obtained from Dolasetron mesylate by solvent and anti-solvent combination process.
- the said process comprises: dissolving Dolasetron mesylate in a polar aprotic solvent at an ambient temperature in the range of 20-35 0 C, preferably in the range of 25-30 0 C, adding an anti-solvent at a temperature in the range of
- the polar aprotic solvent is selected as dissolution solvents.
- the polar aprotic solvent is N, N-dimethyl acetamide.
- the anti-solvent is selected from cyclic ethers such as 1, 4-dioxane.
- Form VII exhibits following peaks:
- the polymorphic Form VIII is obtained by suspending Dolasetron mesylate in aliphatic ketones such as ethyl methyl ketone, heating at a temperature in the range of 30- 85°C for one hour, preferably in the range of 75-8O 0 C, stirring the solution with cooling at a temperature in the range of -5 to 30 0 C, preferably in the range of 25-30 0 C, isolating and drying the product at a temperature in the range of in the range of 40-90 0 C, preferably in the range of in the range of 65-7O 0 C.
- the XRPD of Dolasetron mesylate Form VIII is given in Figure 8.
- the XRPD of Dolasetron mesylate Form VIII exhibits following peaks:
- Polymorphic Form IX of Dolasetron mesylate is obtained from Dolasetron mesylate by solvent and anti-solvent combination process.
- the solvent used for dissolution is selected from lower aliphatic alcohols.
- the lower alcohol is selected from methanol, ethanol and n-propanol.
- the anti-solvent is selected from a group of lower aliphatic ethers.
- the lower aliphatic ether is selected from diethyl ether, diisopropyl ether, and methyl tert. butyl ether.
- the XRPD of Dolasetron mesylate Form IX is given in Figure 9.
- the XRPD of Dolasetron mesylate Form IX exhibits following peaks:
- Amorphous form of Dolasetron mesylate is obtained by lyophilization or vacuum evaporation or by spray drying.
- the solution of Dolasetron mesylate in polar protic solvents is subjected to lyophilization or vacuum evaporation or spray drying to obtain the amorphous form.
- the polar protic solvent used for dissolution is lower alcohols or water.
- the lower alcohols are selected from methanol, ethanol, and n-propanol.
- melt crystallization comprising: melting Dolasetron mesylate at a temperature range of 150-170 0 C, preferably in the range of 160-165 0 C and cooling the melt at a temperature range of 25-45°C, preferably in the range of 25-3O 0 C.
- the XRPD of amorphous Form is given in Figure 10.
- the crystallization process hitherto described to prepare the novel polymorphs comprises, dissolving Dolasetron mesylate in the selected solvent either with or without heating, preferably with heating at or near boiling point of the solvent.
- the resultant solution is cooled to — 5°C to 30 0 C for several hours to regenerate the solid.
- the precipitated solids are isolated and dried at about ambient to 65°C temperature.
- the solvent and anti-solvent combination process described to prepare the novel polymorphs comprises dissolving Dolasetron mesylate in the selected solvent.
- the dissolution is carried out at room temperature or under reflux condition.
- Anti-solvent is added to the resulting solution under warm conditions to regenerate Dolasetron mesylate.
- the anti-solvent addition is generally carried out at room temperature or at 35°C-55°C.
- the precipitated solids are isolated and dried at about ambient to 65 0 C temperature.
- Treatment process described hitherto to prepare Form VIII comprises, suspending Dolasetron mesylate in the selected solvent, refluxing the suspension for 1 hr, and cooling the suspension to -5°C to 30° C, preferably to room temperature under stirring for 3 hr.
- the solids are isolated and dried at about ambient to 65 0 C temperature to obtain crystalline Form VIII.
- the melt crystallization technique described to prepare amorphous form comprises heating of Dolasetron mesylate to form a melt.
- the heating is generally carried out at temperature below 175 0 C.
- the melt of Dolasetron mesylate is generally formed in the temperature range of 150 0 C -175°C, preferably 16O 0 C.
- the melt is allowed to solidify at -5°C to 30 0 C to provide the novel amorphous form.
- the vacuum evaporation technique described to prepare amorphous form consists of evaporation of the solvent from Dolasetron mesylate solution under vacuum.
- the spray drying technique described to prepare amorphous form Dolasetron mesylate consist of aspirating the solution of Dolasetron mesylate at the inlet temperature range of 120 0 C to 180 0 C, preferably 155°C- 165°C and outlet temperature range of 60 0 C to 110 0 C, preferably 95°C -105 0 C.
- the lyophilisation technique described to prepare amorphous form consists of freeze drying an aqueous solution of Dolasetron mesylate.
- the novel polymorphs of Dolasetron mesylate are characterized by X-ray powder diffraction.
- Example 3 Preparation of ⁇ -ethoxycarbonylglutaraldehyde (8)
- a suspension of periodic acid (1.5 Kg, 6.58 mole) in ethyl acetate (3 L) was stirred at 0-10 0 C under nitrogen atmosphere.
- l-ethoxycarbonyl-3- cyclopenteneoxide (19) (1 Kg, 6.40 mole) in ethyl acetate (3 L) in a drop wise manner at 0-10 0 C for lhr.
- the reaction mixture was stirred at 0-10 0 C for 4 hr.
- the reaction mixture was filtered through celite. The filtrate was washed with water (2 x 750 mL).
- reaction mixture was extracted with ethyl acetate (3.0 L), the ethyl acetate layer was separated, washed with water and concentrated to obtain endo-hexahydro-8-(t-butyldimethylsilyloxy)-2-ethoxycarbonyl-2,6-methano-2H- quinolizin-3-(4H)-one(21). Yield: 270 g, 92.15%.
- Example 10 Preparation of endo-hexahydro-8-hydroxy-2,6-methano-2H-quinolizin- 3-(4H)-one (11)
- the reaction mixture was refluxed for 16 hr and cooled to room temperature and basified with potassium carbonate till pH becomes 8-8.5. This solution was saturated with sodium chloride and extracted with isopropanol.
- EXAMPLE 11 Preparation of endo-hexahydro-8-(3-indolylcarbonyloxy)-2,6- methano-2H-quinoIizin-3(4H)-one (Dolasetron base).
- a solution of trifluoroacetic anhydride (413.7 g, 1.97 mole) in dichloromethane (1700 mL) was stirred under nitrogen atmosphere and to this, indole-3-carboxylic acid (302 g, 1.87 moles) was added in a portion wise manner for 30 min at -5 to O 0 C. The reaction mixture was stirred further 30 min at -5 to 0 0 C.
- Step VII endo-hexahydro-8- hydroxy-2,6-methano-2H-quinolizin-3-(4H)-one (Step VII) (170 g, 0.939 moles) in dichloromethane (850 mL) was added in a drop wise manner for 30 min at —5 to 0 0 C and was added dimethyl amino pyridine (1.43 g). The reaction mixture was stirred further for 12 h at room temperature. The reaction mixture was filtered and the collected solid washed with dichloromethane (3 x 170 mL). The solid was stirred in water (2550 ml) and 10% sodium carbonate (1360 mL) for 30 min. The solid formed was filtered and washed with water.
- Example 12 Preparation of endo-hexahydro-8-(3-indoly ⁇ carbonyloxy)-2,6- methano-2H-quinolizin-3(4H)-one (Dolasetron base) A solution of trifluoroacetic anhydride (121.8 g, 0.57 mole) in dichloromethane
- Dolasetron base 50 g, 0.15 mole was dissolved in acetone (1000 mL) and methane sulphonic acid was added (10.70 mL) drop wise over a period of 30 min at 20 0 C. The reaction mixture was stirred further for 2 hr. The solid formed was filtered, washed with cold acetone (50 mL) and dried. Yield (crude) 59 g, 90.77%.
- Example 14 Purification of endo-hexahydro-8-(3-indolylcarbonyIoxy)-2,6- methano-2H-quinolizin-3(4H)-one mesylate, Dolasetron mesylate.
- Step VIII To Dolasetron base (119 g, 0.368 moles) (Step VIII) was dissolved in acetone (2023 mL) and treated with activated charcoal (12 g). Filtered the mixture through hyflow and the clear solution was treated with water (24 ml) and methane sulphonic acid (38.96 g, 0.405 moles) at 25-30°C. The reaction mass was stirred further for 2 h at 0-5 0 C. The solid formed was filtered, washed with acetone (3 x 120 mL) and dried. Yield (crude) 140 g, 87%.
- Example 16 Purification of endo-hexahydro-8-(3-indoIylcarbonyloxy)-2,6- methano-2H-quinolizin-3(4H)-one mesylate, Dolasetron mesylate hydrate.
- the Dolasetron mesylate (140 g) (Step IX) was taken in water (900 ml) and extracted with ethyl acetate (3x280 ml). The aqueous layer was separated, basified with 10% sodium carbonate (320 mL). The solid obtained was filtered, washed with water and dried. This solid was dissolved in acetone (2 xlOO mL) and treated with activated charcoal (12 g). Filtered the mixture through hyfiow and clear solution was treated with water (20 mL) and methane sulphonic acid (32.72 g, 0.341 moles) at 25-30 0 C. The reaction mass was stirred further for 2 h at 0-5 0 C. The solid formed was filtered, washed with acetone (3 xlOO niL) and dried. Yield 130 g, 93%. Purity: 99.9% (HPLC).
- Example 24 0.5g of Dolasetron mesylate was dissolved in 2 mL of DMSO at room temperature.
- Dolasetron mesylate 0.5g was dissolved in 75 mL of chloroform at reflux temperature. The hot solution was maintained at the same temperature for 30 min. The hot solution was allowed to cool to room temperature and was stirred for 3 hr at the same temperature. The solid obtained was filtered and dried at 65 0 C to get Dolasetron mesylate
- Example 35 0.5g of Dolasetron mesylate was dissolved in 75 mL of methylene dichloride at reflux temperature. The hot solution was maintained at the same temperature for 30 min.
- a 5OmL aqueous solution of Dolasetron mesylate at a concentration of 20% weight/volume and at a temperature of 30 0 C was spray dried by a spray gun (PSD 00 Pilot, Hemraj, India at pressure 500 to 600 psi and flow rate of 2 L/hr) at an inlet temperature of 165 C and outlet temperature of 105 C of the spray gun.
- PSD 00 Pilot, Hemraj, India at pressure 500 to 600 psi and flow rate of 2 L/hr
- Example 45 The procedure of Example 45 was carried out at inlet temperature of 155 C and outlet temperature of 95 0 C of the spray gun.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
The present disclosure relates to novel crystalline polymorphs, Form II, III, IV, V, VI, VII, VIII and IX of Dolasetron mesylate and industrial processes for producing them. Further, it discloses processes for producing Form I of Dolasetron mesylate. Furthermore, it relates to the novel amorphous form of Dolasetron mesylate and industrial processes for producing it.
Description
POLYMORPHIC FORMS OF DOLASETRON MESYLATE AND PROCESSES
THEREOF
This specification claims priority from 1635/MUM/2005 dt 29/12/2005 and 1610/MUM/2005 dt 23/12/2005 TECHNICAL FIELD
The present disclosure relates to novel crystalline polymorphs of Dolasetron mesylate having formula (1) and industrial processes for producing the same. Further, it discloses processes for producing Form I of Dolasetron mesylate. Furthermore, the present disclosure teaches novel amorphous form and industrial processes for producing amorphous form.
3H

Formula (1) BACKGROUND AND PRIOR ART Dolasetron mesylate is an antinauseant and antiemetic agent. It is a selective serotonin 5-HT3 receptor antagonist and is indicated for the prevention of nausea and vomiting associated with emetogenic cancer chemotherapy.
Synthesis of Dolasetron mesylate is not very widely reported in literature. EP0266730/US4906755 describes process for the preparation endo-hexahydro-8-(3- indolylcarbonyloxy)-2,6-methano-2H-qumolizin-3(4H)-one methanesulfonate or Dolasetron mesylate (1) by the condensation of diethyl malonate with cis-l,4-dichloro-2- butene (2) in presence of lithium hydride in dimethylformamide to give diethyl-3- cyclopentene-l,l-dicarboxylate (3), which on hydrolysis and decarboxylation gave 3- cyclopentene-1-carboxylic acid (4). The compound (4) was further treated with thionyl chloride and pyridine in ethanol to obtain ethyl 3-cyclopentene-l-carboxylate (5). Compound (5) was oxidized to 4-ethoxycarbonyl-l, 2-cyclopentanediol (6) by using N- methylmorpholine N-oxide in the presence of osmium tetroxide catalyst. The diol (6) was cleaved to the /3-ethoxycarbonylglutaraldehyde (7) using sodium periodate and used, directly in the next reaction. Robinson-Schopf cyclisation of the compound (7) with potassium hydrogen phthalate, acetonedicarboxylic acid and glycine ethyl ester
hydrochloride resulted in the pseudopelletierine derivative i.e. 7-ethoxycarbonyl-9- (ethoxycarbonylmethyl)-9-azabicyclo-[3.3.1]nonan-3-one (8). The ketone group of compound (8) was reduced with sodiumborohydride in ethanol to give 7-ethoxycarbonyl- 9-(ethoxycarbonylmethyl)-9-azabicyclo-[3.3.1]nonan-3-ol (9). The reduced alcohol (9) was treated with dihydropyran to protect the hydroxyl group as a tetrahydropyranyl ether (10). Dieckmann cyclisation of the compound (10) using strong base (potassium t- butoxide) followed by aqueous acid hydrolysis and decarboxylation gave the desired alcohol. The resulting alcohols can exist in two conformations - axial and equatorial. The main product obtained by above procedure was the axial alcohol or endo-hexahydro-8- hydroxy-2,6-methano-2H-quinolizin-3-(4H)-one (11) and it can be separated from the equatorial isomer by crystallization of the camphorsulfonate or tetrafluoroborate salt. The tetrafluoroborate salt of endo-hexahydro-8-hydroxy-2,6-methano-2H-quinolizin-3-(4H)- one (11) was further reacted with 3-indolecarboxylic acid chloride in presence of silver tetrafluoroborate in anhydrous nitroethane at -780C to endo-hexahydro-8-(3- indolylcarbonyloxy)-2,6-methano-2H-quinolizin-3(4H)-one or Dolasetron base, which was further converted into Dolasetron mesylate monohydrate (Scheme I) with a yield of 66%. No further purification is described.
The above process uses column chromatography for purification of compounds (9) and (10), which is expensive, time consuming and impractical on an industrial scale. The above patent does not disclose the yield and purity of Dolasetron mesylate obtained and so also for the intermediates. In addition, Osmium tetroxide used for preparation of compound (6) is toxic, has a corrosive action on eyes and hence difficult to use at industrial scale. Also, this process uses high volume of water during preparation of the compound (8); preparation of compound (11) from compound (10) is tedious, because the workup involves several extractions with ethyl acetate and preparation of compound (1) in presence of silver tetrafluoroborate involves the use of expensive silver compound.
SCHEME I
(2) (3) (4)
COOEt

(5) (6) (7)
CH2COOEt CH2COOEt

(8) (9)
CH COOEt

(10) (H)

Another method described in EP0339669 provides a process for the preparation of endo-hexahydro-8-(3-indolylcarbonyloxy)-2,6-methano-2H-quinolizin-3(4H)-one methanesulfonate or Dolasetron mesylate (1) by the condensation of dimethyl malonate with cis-l,4-dichloro-2-butene (2) in presence of lithium hydride in dimethyl formamide to give dimethyl-3-cyclopentene-l,l-dicarboxylate (12), which was decarboxymethylated to obtain methyl-3-cyclopentene-l-carboxylate (13). This compound (13) was treated with m-chloroperbenzoic acid in dichloromethane to obtain l-methoxycarbonyl-3- cyclopenteneoxide (14). The compound (13) on ozonolysis gave β-
methoxycarbonylglutaraldehyde (15) or the epoxide (14) was reacted with periodic acid to obtain the β-methoxycarbonylglutaraldehyde (15), which was used directly in the next reaction. Robinson-Schopf cyclisation of the compound (15) with potassium hydrogen phthalate, acetonedicarboxylic acid and glycine ethyl ester hydrochloride gave the pseudopelletierine derivative i.e. 7-methoxycarbonyl-9-(methoxycarbonylmethyl)-9- azabicyclo [3.3.1] nonan-3-one (16). The ketone group of compound (16) was reduced with sodiumborohydride in methanol to give 7-methoxycarbonyl-9- (methoxycarbonylmethyrj-P-azabicyclo-p.S.lJnonan-S-ol (17). The reduced alcohol (17) was treated with dihydropyran to protect the hydroxyl group as a tetrahydropyranyl ether (18a) or treated with methylal to protect the hydroxyl group to obtain 3-methoxymethoxy- 7-methoxycarbonyl-9-(methoxycarbonylmethyl)-9-azabicyclo[3.3.1]nonan-3-ol (18b).
Dieckmann cyclisation of the compound (18) using strong base (potassium t- butoxide) followed by aqueous acid hydrolysis and decarboxylation gave the endo- hexahydro-8-hydroxy-2, 6-methano-2H-quinolizin-3-(4H)-one (11). The alcohol (11) was further reacted with 3-indolecarboxylic acid in presence of trifluoroacetic anhydride in dichloromethane to endo-hexahydro-8-(3-indolylcarbonyloxy)-2, 6-methano-2H- quinolizin-3(4H)-one or Dolasetron base, which was then converted into Dolasetron mesylate (1) (Scheme II) by treating with methanesulphonicacid in acetone. Further, crude Dolasetron mesylate (1) was dissolved in aqueous isopropanol and regenerated by adding ether to obtain Dolasetron mesylate (1) with a yield of 85.90 %. Disadvantages of this process are:
(i) use of high volume of water for preparation of compound (16) and (ii) preparation of compound (11) from compound (18) which is tedious because at the time of workup, ethyl acetate extractions take up longer period (20 hr); The process is not only time consuming but also expensive on an industrial scale.
The patent does not disclose purity of Dolasetron obtained nor for any of the intermediates.
SCHEME II

(18)
(11)
ISa. R = I
O
D D
18b. R = CH2OCH3

The process as described in EP 0266730 involves treatment of endo-hexahydro-8- (3-indolylcarbonyloxy)-2, 6-methano-2H-quinolizin-3(4H)-one with a solution of methane sulfonic acid in ethanol to provide Dolasetron mesylate monohydrate. EP 0339669 describes crystallization of crude Dolasetron mesylate by dissolution in aqueous isopropanol and regeneration by adding ether. The polymorphic form obtained by the processes described in US 4906755/EP 0266730 and EP 0339669 is designated herein as Dolasetron mesylate Form I. XRPD of Dolasetron mesylate Form I is disclosed in Figure 1.
The ability of the compound to exhibit more than one orientation or conformation of molecule within the crystal lattice is called polymorphism. Many organic compounds including active pharmaceutical ingredients (API's) exhibit polymorphism.
Drug substance existing in various polymorphic forms differs from each other in terms of stability, solubility, compressibility, flowability and spectroscopic properties, thus affecting dissolution, bioavailability and handling characteristics of the substance.
Rate of dissolution of an API's in patient's stomach fluid can have therapeutic consequences since it imposes an upper limit on the rate at which an orally administrated API can reach the patient . bloodstream. Flowability affects the ease with which the material is handled while processing a pharmaceutical product.
Investigation of crystal polymorphism is an essential step in pharmaceutical research due to the influence of solid-state properties on dosage form.
As the polymorphs are known to possess different spectroscopic properties, technique such as X-Ray powder diffraction (XRPD), Fourier transformer Infrared (FT- IR) spectroscopy, Solid State 13C-NMR, and thermal method of analysis are keys to identify and characterize the new polymorphs or existing polymorphs.
The discovery of new polymorphs with same or better pharmaceutical equivalence and bioequivalence as that of the known polymorphs provides an opportunity to improve the performance characteristic of the pharmaceutical product. Polymorphs of Dolasetron mesylate are not widely reported. CN 1629161 discloses a crystalline polymorph of Dolasetron mesylate monohydrate. In our endeavour to develop a process for the purification of Dolasetron mesylate, we have surprisingly discovered novel polymorphic forms Dolasetron mesylate.
OBJECTS OF THE PRESENT INVENTION
It is an object of the present disclosure to provide novel polymorphic forms of Dolasetron mesylate and industrial processes for producing them.
Another object is to provide a process for preparation of Dolasetron mesylate polymorphic Form I. It is also an object of to provide novel amorphous form of Dolasetron mesylate and industrial processes for producing it. SUMMARY OF THE INVENTION
Accordingly, the present disclosure provides a process for the preparation of a crystalline polymorphic Form I of endo-hexahydro-8-(3-indolylcarbonyloxy)-2, 6- methano-2H-quinolizin-3 (4H)-one methanesulfonate (Dolasetron rrfesyϊate).
In one aspect, the present invention provides a crystalline polymorphic Form II of endo-hexahydro-8-(3-indolylcarbonyloxy)-2,6-methano-2H-quinolizm-3(4H)-one methanesulfonate, Dolasetron mesylate.
In another aspect, the present invention provides a process for producing, polymorphic Form II of Dolasetron mesylate.
In one aspect, the present invention provides a crystalline polymorphic Form III of endo-hexahydro-8-(3-indolylcarbonyloxy)-2,6-methano-2H-quinolizin-3 (4H)-one methanesulfonate, Dolasetron mesylate.
In another aspect, the present invention provides a process for producing polymorphic Form III of Dolasetron mesylate.
In one aspect, the present invention provides a crystalline polymorphic Form IV of endo-hexahydro-8-(3-indolylcarbonyloxy)-2,6-methano-2H-quinolizin-3(4H)-one methanesulfonate, Dolasetron mesylate.
In a further aspect, the present invention relates to a process for producing polymorphic Form IV of Dolasetron mesylate.
In one aspect, the present invention provides a crystalline polymorphic Form V of endo-hexahydro~8-(3 -indolylcarbonyloxy)-2,6-methano-2H-quinolizin-3 (4H)-one methanesulfonate, Dolasetron mesylate.
In another aspect, the present invention provides a process for producing polymorphic Form V of Dolasetron mesylate.
In yet another aspect, the present invention provides a crystalline polymorphic Form VI of endo-hexahydro-8-(3-indolylcarbonyloxy)-2,6-methano-2H-quinolizin- 3(4H)-one methanesulfonate, Dolasetron mesylate.
In a further aspect, the present invention provides a process for producing polymorphic Form VI of Dolasetron mesylate.
In one aspect, the present invention provides a crystalline polymorphic Form VII of endo-hexahydro-8-(3-indolylcarbonyloxy)-2,6-methanό-2H-quinolizin-3(4H)-one methanesulfonate, Dolasetron mesylate.
In another aspect, the present invention provides a process for producing polymorphic Form VII of Dolasetron mesylate.
In yet another aspect, the present invention provides a crystalline polymorphic Form VIII of endo-hexahydro-8-(3-indolylcarbonyloxy)-2,6-methano-2H-quinolizm- 3(4H)-one methanesulfonate, Dolasetron mesylate.
In a further aspect, the present invention provides a process for producing polymorphic Form VIII of Dolasetron mesylate.
In one aspect, the present invention provides a crystalline polymorphic Form IX of endo-hexahydro-8-(3-indolylcarbonyloxy)-2,6-methano-2H-quinolizin-3(4H)-one methanesulfonate, Dolasetron mesylate.
In another aspect, the present invention provides a process for producing polymorphic Form K of Dolasetron mesylate.
In one aspect, the present invention provides an amorphous form of endo- hexahydro-8-(3-indolylcarbonyloxy)-2, 6-methano-2H-quinolizin-3 (4H)-one methanesulfonate, Dolasetron mesylate.
In another aspect, the present invention provides processes for preparation of amorphous form of Dolasetron mesylate.
BRIEF DESCRIPTION OF ACCOMPANYING DRAWINGS
Figure 1 shows XRPD pattern of Dolasetron mesylate Form I
Figure 2 shows XRPD pattern of Dolasetron mesylate Form II
Figure 3 shows XRPD pattern of Dolasetron mesylate Form III Figure 4 shows XRPD pattern of Dolasetron mesylate Form IV
Figure 5 shows XRPD pattern of Dolasetron mesylate Form V
Figure 6 shows XRPD pattern of Dolasetron mesylate Form VI
Figure 7 shows XRPD pattern of Dolasetron mesylate Form VII
Figure 8 shows XRPD pattern of Dolasetron mesylate Form VIII Figure 9 shows XRPD pattern of Dolasetron mesylate Form IX
Figure 10 shows XRPD pattern of amorphous form of Dolasetron mesylate
DETAILED DESCRIPTION OF THE INVENTION
The present disclosure relates to a process for preparation of Dolasetron mesylate or endo-hexahydro-8-(3-indolylcarbonyloxy)-2, 6-methano-2H-quinolizin-3 (4H)-one methanesulfonate of the formula (1) in high yield and high purity comprising:

Formula (1) a. reacting compound (4)

^X^COOR
Formula (V) b. treating the compound haying the structural formula (V) with m-chloroperbenzoic acid in dichloromethane to give an epoxide having the structural formula (XIX),

Formula (XIX) c. treating the epoxide having the structural formula (XIX) with periodic acid to give compound having the structural formula (VII);
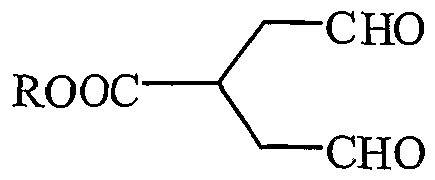
Formula (VII) d. cyclising the compound having the structural formula (VII) with potassium hydrogen phthalate, acetonedicarboxylic acid and glycine ester hydrochloride by Robinson- Schopf cyclisation to obtain pseudopelletierine derivative having the structural formula (VIII);
CH2COOR1

Formula (VIII) e. reducing the compound having the structural formula (VIII) with sodiumborohydride in alcohol followed by treatment with an organic acid to obtain compound having the structural formula (IX);
CH2COOR1

Formula (IX) f. protecting the compound having the structural formula (IX) as a silyl derivative having the structural formula (XX) by treating it with a silyl halide in an organic solvent (wherein Z = silyl group);
CH2COOR1

Formula (XX)
g. treating the compound having the structural formula (XX) with a strong base to form compound having the structural formula (XXI);

Formula (XXI) h. treating the compound having the structural formula (XXI) with acid in water or an organic solvent followed by decarboxylation to yield compound (11);

Compound (11) i. reacting the compound (11) with indole-3-carboxylic acid in presence of trifiuoroacetic acid anhydride to yield Dolasetron base; and j. converting the Dolasetron base into its mesylate; and recrystallizing from a mixture of solvents to obtain highly pure compound (1); wherein, R = Et, Me, or OCH2Ph, Ri = Et, Me, or OCH2Ph and Z is selected from trimethyl silyl, isopropyl dimethyl silyl, t-butyldimethyl silyl, t-butyldiphenyl silyl, tribenzyl silyl, and triisopropyl silyl.
In accordance to above, Scheme III as given below depicts a process for the preparation of 3-cyclopentene-l-carboxylic acid ester (5) is disclosed, said process comprising: reacting 3-cyclopentene-l-carboxylic acid (4) with anhydrous HCl gas or concentrated hydrochloric acid or thionyl chloride in an alcohol, wherein the alcohol is either methanol or ethanol; treating the compound (5) with m-chloroperbenzoic acid in a solvent selected from dichloromethane and ethyl acetate to obtain the corresponding epoxide (19); reacting the compound (19) with periodic acid under nitrogen atmosphere to obtain compound (7); treating the compound (7) with potassium hydrogen phthalate, acetonedicarboxylic acid and glycine ester hydrochloride in water to obtain pseudopelletierine derivative (8); reducing the compound (8) with sodiumborohydride in
an alcohol and further treating with an organic acid to obtain compound (9), wherein the organic acid is selected from formic acid, methane sulphonic acid and acetic acid; treating the compound (9) with silyl halide in presence of imidazole in an organic solvent to obtain compound (20), wherein the organic solvent is selected from ketones, esters and ethers, preferably from acetone, tetrahydrofuran, 1, 4-dioxane, dichloromethane, chloroform, N, N-dimethyl formamide, ethyl acetate and acetonitrile.
Scheme III
(4) (5) (19)

(8) (7) C H2COOEt


(9) (20)

late

(Dolasetron base)
A major advantage of the use of silyl protecting group is that it yields greater than 95 % of compound (20) as compared to, use of dihydropyran (75%) or methylal (84%).
The compound (20) is treated with a strong base in toluene and further treated with an organic acid in an organic solvent to form compound (21). The organic solvent is selected from halogenated solvents, ethers and esters. The organic solvent is preferably selected from methylene chloride, chloroform, ethyl acetate, isopropyl acetate, diethyl
ether, diisopropyl ether or mixtures thereof. The organic acid is selected from formic acid and acetic acid.
The compound (21) is heated with hydrochloric acid in water to give compound (11). Hydrochloric acid and water are used in the ratio of 1:2 volumes. The ratio of compound (21) to water in the reaction is about 1: 8 to 1:10. The reaction mixture is concentrated and the residue obtained is treated with an organic solvent and filtered. The filtrate is concentrated to obtain compound (11). The organic solvent is selected from alcohols and halogenated solvent preferably methanol, ethanol, isopropanol, n-butanol, dichloromethane, chloroform or mixture thereof. The reaction mixture is extracted with an organic solvent selected from ethyl acetate, isopropanol or n-butanol. Alternately the reaction mixture is saturated with an inorganic salt and extracted with an organic solvent selected from ethyl acetate or n-butanol or isopropanol.
The compound (11) is reacted with indole-3-carboxylic acid in presence of trifiuoroacetic acid anhydride in dichloromethane to give Dolasetron base. The ratio of indole-3-carboxylic acid and trifluoro acetic anhydride used is in the range of 1:1.1 to 1:2. Dolasetron base thus obtained is isolated by conventional method. Dolasetron base is solubilized in acetone and converted into its mesylate salt using methane sulphonic acid. The resultant mesylate salt is dissolved in water and extracted with a halogenated solvent or ester to remove traces of impurity. The halogenated solvent is selected from dichloromethane and chloroform, and the ester is selected from methyl acetate, ethyl acetate and isopropyl acetate. The aqueous layer is basified with a base to obtain Dolasetron base. The base is selected from sodium carbonate, potassium carbonate, sodium bicarbonate, potassium bicarbonate, sodium hydroxide, potassium hydroxide or mixtures thereof. Dolasetron base thus obtained is treated with methane sulphonic acid in a mixture of acetone and water to provide Dolasetron Mesylate. Polymorphic forms of Dolasetron mesylate
One more embodiment of the invention provides novel crystalline polymorphic forms, viz: Form II, Form III, Form IV, Form V, Form VI, Form VII, Form VIII and Form IX. Another embodiment of the invention provides a process for manufacturing these crystalline polymorphic forms. The invention also discloses novel amorphous form of Dolasetron mesylate.
The polymorphic Form I of Dolasetron mesylate is obtained by crystallization. The process involves, dissolving Dolasetron mesylate in a solvent selected from aliphatic alcohols, aliphatic ketones, aliphatic esters, aliphatic nitriles or mixtures thereof at a temperature in the range of 30°C-80°C to yield a clear solution. The clear solution is cooled at a temperature in the range of 0°C-30°C, preferably in the range of 25°C-30°C to i obtain a solid. Dolasetron mesylate form I is obtained by filtering and drying the solid at a temperature in the range of 30°C-90°C, preferably in the range of 60°C-70°C. The aliphatic alcohol selected is isopropanol; the aliphatic ester is selected from methyl acetate, ethyl acetate and butyl acetate; aliphatic ketone is selected from acetone, 2- butanone, diethylketone, and the like; and aliphatic nitrile is acetonitrile.
Dolasetron mesylate Form I is also obtained by solvent and anti-solvent process. The said process comprises: dissolving Dolasetron mesylate in a solubilizing solvent, adding an anti-solvent, stirring the suspension with or without cooling, isolating and drying the product at 50°C-70°C. The solubilizing solvents selected for dissolution are polar aprotic solvents. The polar aprotic solvent is selected from N, N-dimethyl formamide, dimethyl sulfoxide and N, N-dimethyl acetamide. The anti-solvent is selected from cyclic ethers, aromatic hydrocarbons and alcohols. The cyclic ether selected is tetrahydrofuran. The aromatic hydrocarbon is toluene and the alcohol is isopropanol.
The XRPD of Dolasetron mesylate Form I exhibit following peaks:
Position [°2Θ] ReI. Int. [%]
7.4587 6.15
9.9618 22.96
10.5229 11.60
11.8311 13.91
12.2113 46.32
12.8014 16.93
13.0334 69.03
13.9997 5.05
14.7702 4.51
15.2057 48.73
15.4577 ' 22.29
16.3397 6.45
16.7613 7.33
17.1871 39.45
18.0049 21.19
18.2978 10.25
19.0431 73.21
19.3804 25.15
19.8238 84.20
20.1208 70.13
20.4718 6.81
21.1534 32.28
22.1965 41.40
22.5131 100.00
22.9364 21.29
23.1046 18.63
23.5992 21.37
24.3924 29.65
24.7468 24.62
25.0691 12.40
25.6221 16.23
26.0495 18.98
26.6353 30.98
27.2251 16.77
28.0475 24.76
28.8492 15.82
29.9349 24.37 .
30.2157 65.62
30.5167 12.70
31.0803 9.84
31.6533 17.87
32.8171 11.68
33.3453 11.07
33.8520 15.00
34.5881 6.22
35.5587 9.61
36.6675 15.29
37.9088 6.97
38.7741 7.86
40.5258 8.59
42.5248 5.86
42.9394 10.15
43.5589 8.97
44.8652 3.63
46.3824 6.20
49.1697 6.31
The polymorphic Form II is obtained by crystallizing Dolasetron mesylate from methanol. The polymorphic Form II is obtained by dissolving Dolasetron mesylate in methanol at a temperature in the range of 30-800C, preferably in the range of 60-700C, cooling the solution at a temperature in the range of -5°C to 250C, preferably in the range of 2 to 70C, isolating and drying the product at a temperature in the range of in the range of 40-900C, preferably in the range of in the range of 60-700C. The XRPD of Dolasetron
mesylate Form II is given in Figure 2. The XRPD of Dolasetron mesylate Form II exhibits following peaks:
Position [°20] ReI. Int. [%]
8.5677 6.09
10.0288 15.17
11.2894 4.56
12.1224 11.85
14.3549 6.44
15.3566 37.98
16.1189 100.00
17.4727 33.33
17.9848 13.61
18.2788 17.61
18.6717 7.36
19.0521 5.06
19.4583 24.09
19.9142 58.36
20.3327 62.55
20.7258 84.65
21.4927 21.14
22.8359 11.20
23.1023 10.08
24.4864 23.47
25.8654 10.96
27.1036 10.58
27.5344 32.07
27.8212 8.51
28.2995 54.08
30.0154 6.14
30.3489 6.50
30.8158 15.55
31.1764 9.57
32.1019 8.14
32.4852 8.13
32.7724 7.66
33.6263 4.20
34.9029 2.52
35.7768 7.29
36.9416 6.79
37.2354 6.99
39.4576 3.12
39.9761 7.25
42.1000 1.20
45.1580 1.23
47.4886 1.74
49.5076 4.99
The polymorphic Form III is obtained by crystallizing Dolasetron mesylate from ethanol. The polymorphic Form III is obtained by dissolving Dolasetron mesylate in ethanol at a temperature in the range of 30-800C, preferably in the range of 75-8O0C, cooling the solution at a temperature in the range of-5°C to 25°C, preferably in the range of 2 to 7°C, isolating and drying the product at a temperature in the range of in the range of 40-900C, preferably in the range of 60-700C.
Also, the polymorphic Form III is obtained by using solvent and anti-solvent combination process. The said process comprises: dissolving Dolasetron mesylate in a solubilizing solvent at a temperature in the range of 30-800C, preferably in the range of 60-800C, adding an anti-solvent at a temperature in the range of 30-550C, preferably in the range of 40-500C, isolating and drying the product at a temperature in the range of 40- 900C, preferably in the range of 60-700C. The solubilizing solvents are selected from lower aliphatic alcohols. The lower alcohol is selected from methanol, ethanol, n-propanol and isopropanol, preferably ethanol. The anti-solvent is selected from aliphatic hydrocarbons n-pentane, n-hexane and n-heptane, preferably n-hexane. The XRPD of Dolasetron mesylate Form III is given in Figure 3. The XRPD of Dolasetron mesylate Form III exhibits following peaks:
Position [°2Θ] ReI. Int. [%]
6.9327 2.87
9.8793 4.08
11.7303 2.68
12.6509 100.00
13.7701 15.97
16.6416 21.33
17.5613 18.64
18.1133 66.76
19.4364 37.96
19.6530 70.74
20.5969 43.14
21.2122 42.37
22.1215 27.17
22.3203 29.08
22.9997 14.64
23.2369 29.45
25.3464 22.52
25.7644 7.93
26.7943 11.14
27.9040 44.38
28.2908 11.30
29.5991 19.08
31.3094 10.72
31.5560 10.97
32.6826 7.96
34.1101 5.85
34.5722 6.62
34.9579 7.12
36.3803 5.26
37.6469 3.01
39.6600 .3.13
40.8489 2.61
41.6942 4.21
44.2459 1.74
45.3305 1.76
Polymorphic Form IV is obtained by crystallizing Dolasetron mesylate from n- propanol. The polymorphic Form IV is obtained by dissolving Dolasetron mesylate in n- propanol at a temperature in the range of 30-1000C, preferably in the range of 90-1000C, cooling the solution at a temperature in the range of -5°C to 250C, preferably in the range of 2 to 7°C, isolating and drying the product at a temperature in the range of in the range of 40-900C, preferably in the range of in the range of 60-700C.
The XRPD of Dolasetron mesylate Form IV is given in Figure 4. The XRPD of Dolasetron mesylate Form IV exhibits following peaks:
Position [°2Θ] ReI. Int. [%]
8.9231 13.20
9.8783 4.58
11.0009 9.65
12.1275 12.74
12.5348 16.42
12.7100 4.78
12.9295 13.19
13.5324 7.67
14.0712 68.91
14.4080 17.98
14.8069 33.77
15.1068 19.18
15.4202 20.16
16.6676 24.07
17.0883 13.11
17.3884 18.93
17.8087 20.51
18.1597 14.53
18.9666 19.40
19.2953 26.33
19.9217 100.00
20.3947 24.69
20.8115 42.34
21.6045 15.47
22.0471 73.52
22.4082 27.53
23.0366 20.15
23.5718 9.97
24.3224 22.76
25.0364 18.40
26.3036 14.18
27.4731 33.17
28.2271 10.21
29.2801 22.56
30.1751 24.53
32.7579 11.03
33.6510 11.10
36.5575 6.71
40.5518 2.52
Polymorphic Form V is obtained by crystallizing Dolasetron mesylate from chlorinated hydrocarbons. The polymorphic Form V is obtained by dissolving Dolasetron mesylate in a solubilizing solvent at a temperature in the range of 30-800C, cooling the solution at a temperature in the range of-5°C to 300C, preferably in the range of 25-3O0C, isolating and drying the product at a temperature in the range of in the range of 40-900C, preferably in the range of in the range of 60-700C. The solubilizing solvent is chlorinated hydrocarbon and is selected from methylene dichloride or chloroform. The XRPD of
Dolasetron mesylate Form V is given in Figure 5. The XRPD of Dolasetron mesylate Form V exhibits following peaks:
Position [°2Θ] ReI. Int. [%]
7.0234 2.39
9.4499 10.44
10.0014 1.75
11.2346 6.13
12.3990 41.17
13.0876 5.00
13.5.899 16.75
14.2116 34.51
14.9901 23.78
16.4007 16.49
16.5997 10.27
17.2415 2.93
18.7313 61.04
19.3572 11.87
19.9122 7.47
20.3109 37.22
20.7126 63.91
21.2339 42.10
22.2956 100.00
23.8661 22.10
24.2519 26.86
24.8389 22.90
25.7209 4.60
26.7060 5.14
27.1085 15.85
27.8334 9.48
28.2062 21.05
28.8706 29.90
29.6637 32.04
30.2901 8.68
31.0608 17.14
31.3191 15.03
33.4700 8.81
34.8387 8.04
35.4257 5.99
36.2944 3.36
39.3031 3.61
40.2044 1.49
40.7189 2.00
41.9681 2.42
42.9023 5.63
44.2007 3.48
45.7069 4.46
48.3668 2.41
Polymorphic Form VI is obtained from Dolasetron mesylate by solvent and anti- solvent combination process. The said process comprises of dissolving Dolasetron mesylate in a solubilizing solvent like a polar aprotic solvent at a temperature in the range of 20-350C, preferably in the range of 25-300C, adding an anti-solvent at a temperature in the range of 20-450C, preferably in the range of 25-300C, isolating and drying the product at a temperature in the range of 40-900C, preferably in the range of 60-700C. The polar aprotic solvent selected for dissolving Dolasetron mesylate is dimethyl formamide or dimethyl sulfoxide. The anti-solvent is selected from cyclic ethers such as 1, 4-dioxane. The XRPD of Dolasetron mesylate Form VI is given in Figure 6. The XRPD of Dolasetron mesylate Form VI exhibits following peaks:
Position [°2Θ] ReI. Int. [%]
7.4433 5.63
9.5049 20.13
9.9496 13.90
10.5039 . 6.65
11.8166 5.42
12.1944 18.96
12.6288 29.00
13.0077 30.68
13.3801 4.62
13.7613 2.83
14.7330 2.72
15.1734 34.41
15.4668 12.92
16.6316 24.00
17.1629 26.82
17.8499 53.38
19.0352 100.00
19.3432 89.80
19.8030 46.30
20.0712 36.98
20.4909 65.99
20.9356 8.65
21.3421 21.25
21.8514 7.35
22.1111 35.66
22.5010 57.85
22.7995 32.62
23.0903 11.37
23.6251 7.97
24.3860 15.83
24.7255 19.47
25.0343 14.83
25.6040 8.62
26.0387 10.14
26.6211 15.13
27.2259 59.06
28.0530 14.52
29.1029 16.69
29.9326 6.76
30.2376 29.39
31.0858 9.70
31.4142 12.45
31.6868 9.26
32.3416 4.26
32.8342 4.78
33.3082 7.59
33.8019 7.03
35.3064 11.79
36.6186 5.25
37.6928 2.32
38.5491 2.80
40.8503 3.65
42.9479 2.85
43.5321 4.78
45.8347 5.25
47.0633 3.28
49.2055 2.24
The polymorphic Foπn VII is obtained from Dolasetron mesylate by solvent and anti-solvent combination process. The said process comprises: dissolving Dolasetron mesylate in a polar aprotic solvent at an ambient temperature in the range of 20-350C, preferably in the range of 25-300C, adding an anti-solvent at a temperature in the range of
20-450C, preferably in the range of 25-300C, isolating and drying the product at a temperature in the range of 30-900C, preferably in the range of 60-700C. The polar aprotic solvent is selected as dissolution solvents. The polar aprotic solvent is N, N-dimethyl acetamide. The anti-solvent is selected from cyclic ethers such as 1, 4-dioxane. The XRPD of Dolasetron mesylate Form VII is given in Figure 7. The XRPD of Dolasetron mesylate
Form VII exhibits following peaks:
Position [°2Θ] ReI. Int. [%]
9.4384 20.32
11.6064 2.06
12.5519 72.26
13.3180 3.73
13.7754 7.33
16.5925 43.77
17.2066 32.10
17.7820 78.62
18.9901 80.48
19.2914 100.00
20.5000 95.46
20.9102 10.74
21.2835 35.90
21.7867 11.90
22.0779 36.86
22.7162 53.61
24.6939 8.55
25.0331 15.90
25.5487 10.34
26.2507 9.33
27.1538 57.00
27.7132 4.84
28.2070 7.44
29.0636 18.19
30.9147 • 10.15
31.3735 14.62
31.7627 9.07
32.3232 4.10
32.9013 5.62
34.0401 7.29
35.2654 13.30
39.1189 3.42
40.7887 3.73
47.0409 3.23
The polymorphic Form VIII is obtained by suspending Dolasetron mesylate in aliphatic ketones such as ethyl methyl ketone, heating at a temperature in the range of 30- 85°C for one hour, preferably in the range of 75-8O0C, stirring the solution with cooling at a temperature in the range of -5 to 300C, preferably in the range of 25-300C, isolating and drying the product at a temperature in the range of in the range of 40-900C, preferably in the range of in the range of 65-7O0C. The XRPD of Dolasetron mesylate Form VIII is given in Figure 8. The XRPD of Dolasetron mesylate Form VIII exhibits following peaks:
Position [°2Θ] ReI. Int. [%]
7,5963 4.24
9.3846 8.81
12.3236 59.04
13.5267 3.90
14.1731 48.06
15.0400 50.64
16.2205 100.00
18.3240 92.38
18.6858 20.79
19.3579 4.00
20.7258 37.10
21.0127 82.31
22.0929 80.73
22.5425 91.43
23.7057 35.67
24.8243 13.17
25.9718 21.22
26.6736 10.77
27.9853 26.04
29.1333 33.12
29.7016 16.86
30.1635 12.14.
31.2731 12.07
32.5971 8.36
33.5183 8.64
34.7327 10.71
37.0768 10.79
45.7888 3.36
Polymorphic Form IX of Dolasetron mesylate is obtained from Dolasetron mesylate by solvent and anti-solvent combination process. The solvent used for dissolution is selected from lower aliphatic alcohols. The lower alcohol is selected from methanol, ethanol and n-propanol. The anti-solvent is selected from a group of lower aliphatic ethers. The lower aliphatic ether is selected from diethyl ether, diisopropyl ether, and methyl tert. butyl ether. The XRPD of Dolasetron mesylate Form IX is given in Figure 9. The XRPD of Dolasetron mesylate Form IX exhibits following peaks:
Position [°2 θ] ReI. Int. [%]
8.9186 5.25
9.5043 13.32
9.9342 8.08
11.3309 10.50
12.2006 27.86
13.0074 25.17
13.5809 26.51
14.2218 66.49
14.7381 39.30
15.1831 41.45
16.5427 21.94
16.9310 24.23
19.0277 29.88
19.7369 88.83
20.3022 100.00
21.2529 23.15
22.5030 75.37
23.1298 ' 18.84
24.3308 15.61
26.5618 14.00
27.3363 28.13
28.3123 62.31
30.1981 35.94
32.6226 11.17
33.8362 12.52
Amorphous form of Dolasetron mesylate is obtained by lyophilization or vacuum evaporation or by spray drying. The solution of Dolasetron mesylate in polar protic solvents is subjected to lyophilization or vacuum evaporation or spray drying to obtain the amorphous form. The polar protic solvent used for dissolution is lower alcohols or water. The lower alcohols are selected from methanol, ethanol, and n-propanol.
Another process to obtain amorphous form is melt crystallization, comprising: melting Dolasetron mesylate at a temperature range of 150-1700C, preferably in the range of 160-1650C and cooling the melt at a temperature range of 25-45°C, preferably in the range of 25-3O0C. The XRPD of amorphous Form is given in Figure 10. The crystallization process hitherto described to prepare the novel polymorphs comprises, dissolving Dolasetron mesylate in the selected solvent either with or without heating, preferably with heating at or near boiling point of the solvent. The resultant solution is cooled to — 5°C to 300C for several hours to regenerate the solid. The precipitated solids are isolated and dried at about ambient to 65°C temperature. The solvent and anti-solvent combination process described to prepare the novel polymorphs comprises dissolving Dolasetron mesylate in the selected solvent. The dissolution is carried out at room temperature or under reflux condition. Anti-solvent is added to the resulting solution under warm conditions to regenerate Dolasetron mesylate. The anti-solvent addition is generally carried out at room temperature or at 35°C-55°C. The precipitated solids are isolated and dried at about ambient to 650C temperature.
Treatment process described hitherto to prepare Form VIII comprises, suspending Dolasetron mesylate in the selected solvent, refluxing the suspension for 1 hr, and cooling the suspension to -5°C to 30° C, preferably to room temperature under stirring for 3 hr. The solids are isolated and dried at about ambient to 650C temperature to obtain crystalline Form VIII.
The melt crystallization technique described to prepare amorphous form comprises heating of Dolasetron mesylate to form a melt. The heating is generally carried out at temperature below 1750C. The melt of Dolasetron mesylate is generally formed in the temperature range of 1500C -175°C, preferably 16O0C. The melt is allowed to solidify at -5°C to 300C to provide the novel amorphous form.
The vacuum evaporation technique described to prepare amorphous form consists of evaporation of the solvent from Dolasetron mesylate solution under vacuum.
The spray drying technique described to prepare amorphous form Dolasetron mesylate consist of aspirating the solution of Dolasetron mesylate at the inlet temperature range of 1200C to 1800C, preferably 155°C- 165°C and outlet temperature range of 600C to 1100C, preferably 95°C -1050C. The lyophilisation technique described to prepare amorphous form consists of freeze drying an aqueous solution of Dolasetron mesylate.
The novel polymorphs of Dolasetron mesylate are characterized by X-ray powder diffraction. X-ray powder diffraction pattern has been obtained on Xpert'PRO, Panalytical diffractometer equipped with accelerator detector using Copper Kce (λ= 1.5406 A) radiation with scanning range between 4-50° 2Θ at a scanning speed of 2°/min.
The present invention is described herein below with examples, which are illustrative only and should not be construed to limit the scope of the present invention in any manner. EXAMPLES Example 1: Preparation of ethyl-S-cyclopentene-l-carboxylate (5)
A solution of 3-cyclopentene-l-carboxylic acid (500 g, 4.45 mole) in ethanol (500 niL) was stirred at 5-100C. Then thionyl chloride (257.59 g, 2.16 mole) was added in a drop wise manner for 1 hr. After complete addition was over, the reaction mixture was stirred at room temperature for 30 min. The reaction mixture was poured into the water (1000 mL) and extracted with ethyl acetate (2 x 250 mL). The ethyl acetate layer was washed with 10% sodium carbonate solution (500 mL), with water (2 x 500 mL) and concentrated to give ethyl-3-cyclopentene-l-carboxylate (5). Yield: 558g, 89.42%. Example 2: Preparation of l-ethoxycarbonyl-S-cyclopenteneoxide (19) A solution of ethyl-3-cyclopentene-l-carboxylate (5) (1 Kg, 7.13 mole) in dichloromethane (8 L) was stirred at 5-100C. Then 70 % meta-chloroperbenzoic acid (2.4 Kg, 9.73 mole) was added in lots for 1 hr at 5-100C. The reaction mixture was stirred at 5- 100C for 3 hr. The reaction was monitored using gas chromatography. The reaction mixture was filtered and cake washed with dichloromethane (2 x 1 L). The filtrate was washed with 10 % sodium metabisulphite (5 L), 10 % sodium carbonate (10 L), dried over sodium sulphate and concentrated to give l-ethoxycarbonyl-3-cyclopenteneoxide (19). Yield: 1.1 Kg, 98.74%. Example 3: Preparation of β-ethoxycarbonylglutaraldehyde (8)
A suspension of periodic acid (1.5 Kg, 6.58 mole) in ethyl acetate (3 L) was stirred at 0-100C under nitrogen atmosphere. Then was added l-ethoxycarbonyl-3- cyclopenteneoxide (19) (1 Kg, 6.40 mole) in ethyl acetate (3 L) in a drop wise manner at 0-100C for lhr. The reaction mixture was stirred at 0-100C for 4 hr. The reaction mixture was filtered through celite. The filtrate was washed with water (2 x 750 mL). The ethyl acetate layer was diluted with water (3 L). From this mixture ethyl acetate was evaporated at 30-350C under vacuum and aqueous layer that remained contained β- ethoxycarbonylglutaraldehyde (7). This aqueous solution was directly used in the next step. Example 4: Preparation of 7-ethoxycarbonyl-9-(ethoxycarbonylmethyl)-9- azabicyclo- [3.3.1]nonan-3-one (8)
A suspension of potassium hydrogen phthalate (2.5 Kg, 12.24 mole) in water (2 L) was stirred at room temperature. Then acetonedicarboxylic acid (1.15 Kg, 8.23 mole) in water (1.4 L) and glycine ethyl ester (1.15 Kg, 8.23 mole) in water (1.6 L) were added to the reaction mixture at 15-200C. The aqueous solution containing β-ethoxycarbonyl glutaraldehy.de (7) was added in a drop wise manner for 1 hr under nitrogen atmosphere. The reaction mixture was stirred for 12 hr at room temperature and the pH was adjusted to 8-8.5 by the addition of the potassium carbonate and extracted with ethyl acetate (3 x lOOO mL). The ethyl acetate layer was separated, washed with water and concentrated to give 7- ethoxycarbonyl-9-(ethoxycarbonylmethyl)-9-azabicyclo-[3.3.1]nonan-3-one (8). Yield: 1.05 Kg, 55.14%.
Example 5: Preparation of 7-ethoxycarbonyl-9-(ethoxycarbonylmethyI)-9- azabicyclo-[3.3.1]nonan-3-ol (9) To a solution of 7-ethoxycarbonyl-9-(ethoxycarbonylmethyl)-9-azabicyclo-
[3.3.1]nonan-3-one (8) (450 g, 1.51 mole) in ethanol (4.5 L) was added, sodiumborohydride (175 g, 4.62 mole) in a portion wise manner for 30 min at 10-150C. The reaction mixture was stirred at room temperature for 2 hr and the pH was adjusted to 7 by the addition of the acetic acid. The solid was filtered and the filtrate was concentrated to yellow residue. Water (1.2 L) was added to the residue and the reaction mixture was basified using 10% potassium carbonate solution and extracted with ethyl acetate (3 x 600 mL). The ethyl acetate layer was separated and concentrated to give 7-ethoxycarbonyl-9- (ethoxycarbonylmethyl)-9-azabicyclo-[3.3.1]nonan-3-ol (9). Yield: 365 g, 80.56%.
Example 6: Preparation of 3-tertiary-butyl dimethylsilyloxy-7-ethoxycarbonyl-9- (ethoxycarbonylmethyty-P-azabicyclo-β.S.llnonan-S-ol (20)
A solution of 7-ethoxycarbonyl-9-(ethoxycarbonylmethyl)-9-azabicyclo- [3.3.1]nonan-3-ol (9) (351 g, 1.17 mole), imidazole (239 g, 3.51 mole) and t- butyldimethylsilyl chloride (265 g, 1.7 mole) in N,N-dimethylformamide (700 mL) was stirred at 100C for 30 min. The reaction mixture was stirred at room temperature for 2 hr, after which it was poured into water (5 L) and extracted with ethyl acetate (3 χ 500 ml). The ethyl acetate layer was separated, washed with water (3 xlOOO mL) and concentrated to give 3-tertiary butyl dimethylsilyloxy 7-ethoxycarbonyl-9- (ethoxycarbonylmethyl)-9-azabicyclo-[3.3.1]nonan-3-ol (20). Yield: 480 g, 99.17%. 1HNMR: 200MHz, CDCl3; the chemical shifts expressed are in δ. 0.1 (s, 6H, 2 x CH3); 0.93 (m, 15H, 5 x CH3); 4.1 to 4.26 (m, 4H, 2 x CH2); 1.27 to 3.47 (m, 13H, 5 x CH2 + 3 x CH).
Example 7: Preparation of endo-hexahydro-8-(t-butyIdimethylsiIyloxy)-2- ethoxycarbonyl-2,6-methano-2H-quinoIizin-3-(4H)-one (21)
A mixture of 3-(t-butyldimethylsilyloxy)-7-ethoxycarbonyl-9-
(ethoxycarbonylmethyl)-9-azabicyclo-[3.3.1]nonan-3-ol (20) (480 g, 1.16 mole) and potassium t-butoxide (235 g, 2.09 mole) in toluene (4.5 L) was refluxed under nitrogen atmosphere for 2 hr. Acetic acid (140 mL) was added to the reaction mixture at 10-150C followed by water (500 mL). The reaction mixture was extracted with ethyl acetate (3.0 L), the ethyl acetate layer was separated, washed with water and concentrated to obtain endo-hexahydro-8-(t-butyldimethylsilyloxy)-2-ethoxycarbonyl-2,6-methano-2H- quinolizin-3-(4H)-one(21). Yield: 270 g, 92.15%.
1HNMR: 200MHz, CDCl3; the chemical shifts expressed are in δ.
0.08 (s, 6H, 2 x CH3); 0.89 (m, 12H, 4 x CH3); 4.1 to 4.23 (m, 4H, 2 x CH2); 1.23 to 4.2
& 4.81 to 5.3 (m, 12H, 5 x CH2 + 2 x CH).
Example 8: Preparation of endo-hexahydro-8-hydroxy-2,6-methano-2H-quinolizin- 3-(4H)-one (11)
To the oily compound, endo-hexahydro-8-(t-butyldimethylsilyloxy)-2- ethoxycarbonyl-2,6-methano-2H-quinolizin-3-(4H)-one (21) (100 g, 0.39 mole) in water
(200 mL) concentrated hydrochloric acid (50 mL) was added. The reaction mixture was refluxed for 16 hr, cooled to room temperature and basified with potassium carbonate till pH becomes 8-8.5. This solution was concentrated under reduced pressure to obtain a residue. This residue was treated with 50% methanol in dichloromethane to precipitate inorganic material. This inorganic material was separated by filtration and filtrate was concentrated to give endo-hexahydro-8-hydroxy-2,6-methano-2H-quinolizin-3-(4H)-one (11). Yield: 26 g, 36.34%.
Example 9: Preparation of endo-hexahydro-8-hydroxy-2,6-methano-2H-quinolizm- 3-(4H)-one (11)
To the oily compound, endo-hexahydro-8-(t-butyldimethylsilyloxy)-2- ethoxycarbonyl-2,6-methano-2H-quinolizin-3-(4H)-one (21) (100 g, 0.39 mole) in water (200 mL) was added concentrated hydrochloric acid (50 mL). The reaction mixture was refluxed for 16 h cooled to room temperature and basified with potassium carbonate till pH becomes 8-8.5. This solution was extracted with n-butanol. The butanol layer was separated and concentrated under reduced to give endo-hexahydro-8-hydroxy-2,6- methano-2H-quinolizin-3-(4H)-one (11). Yield: 25.5 g, 35.64%.
Example 10: Preparation of endo-hexahydro-8-hydroxy-2,6-methano-2H-quinolizin- 3-(4H)-one (11) To the oily compound endo-hexahydro-8-(t-butyldimethylsilyloxy)-2- ethoxycarbonyl-2,6-methano-2H-quinolizin-3-(4H)-one (21) (100 g, 0.39 mole) in water (200 mL) was added concentrated hydrochloric acid (50 mL). The reaction mixture was refluxed for 16 hr and cooled to room temperature and basified with potassium carbonate till pH becomes 8-8.5. This solution was saturated with sodium chloride and extracted with isopropanol. The isopropanol layer was separated and concentrated under reduced pressure to give residue. This residue was treated with dichloromethane and clear solution of dichloromethane was filtered and concentrated to provide endo-hexahydro-8-hydroxy- 2,6-methano-2H-quinolizin-3-(4H)-one (11). Yield: 25.5 g, 35.64%.
EXAMPLE 11: Preparation of endo-hexahydro-8-(3-indolylcarbonyloxy)-2,6- methano-2H-quinoIizin-3(4H)-one (Dolasetron base).
A solution of trifluoroacetic anhydride (413.7 g, 1.97 mole) in dichloromethane (1700 mL) was stirred under nitrogen atmosphere and to this, indole-3-carboxylic acid (302 g, 1.87 moles) was added in a portion wise manner for 30 min at -5 to O0C. The reaction mixture was stirred further 30 min at -5 to 00C. Then endo-hexahydro-8- hydroxy-2,6-methano-2H-quinolizin-3-(4H)-one (Step VII) (170 g, 0.939 moles) in dichloromethane (850 mL) was added in a drop wise manner for 30 min at —5 to 00C and was added dimethyl amino pyridine (1.43 g). The reaction mixture was stirred further for 12 h at room temperature. The reaction mixture was filtered and the collected solid washed with dichloromethane (3 x 170 mL). The solid was stirred in water (2550 ml) and 10% sodium carbonate (1360 mL) for 30 min. The solid formed was filtered and washed with water. This solid was stirred with 5 % methanesulphonic acid (850 mL) for 1 h and filtered to remove excess undissolved indole 3-carboxylic acid. The filtrate was extracted with ethyl acetate (3 x 340 ml) and the ethyl acetate layer was separated. The aqueous acidic layer was basified with 10% sodium carbonate (850 mL), solid was separated, filtered and washed with water. Dried the wet solid (Dolasetron base). Yield: 127 g, 42%.
Example 12: Preparation of endo-hexahydro-8-(3-indolyϊcarbonyloxy)-2,6- methano-2H-quinolizin-3(4H)-one (Dolasetron base) A solution of trifluoroacetic anhydride (121.8 g, 0.57 mole) in dichloromethane
(750 mL) was stirred under nitrogen atmosphere and to this, indole-3-carboxylic acid (88 g, 0.54 mole) was added in a portion wise manner for 30 min at 0 to 50C, The reaction mixture was stirred for further 30 min at 0-50C. Then endo-hexahydro-8-hydroxy-2,6- methano-2H-quinolizin-3-(4H)-one (11) (50 g, 0.27 mole) in dichloromethane (500 mL) and dimethyl amino pyridine (0.42 g, 0.0039 mole) were added in a drop wise manner for 30 min at 0-50C. The reaction mixture was stirred further for 12 h at room temperature. The reaction mixture was filtered and the collected solid washed with dichloromethane (100 mL). The solid was stirred in ethyl acetate (550 mL) and 10% sodium carbonate (500 mL) was further added. The ethyl acetate layer was separated, washed with water and concentrated to obtain crude "Dolasetron base (60 g). The crude base was recrystallized from ethyl acetate-hexane to give pure Dolasetron base. Yield: 50 g, 50.63%.
Example 13: Preparation of endo-hexahydro-8-(3-indolylcarbonyloxy)-2,6- methano-2H-quinoIizin-3(4H)-one mesylate
Dolasetron base (50 g, 0.15 mole) was dissolved in acetone (1000 mL) and methane sulphonic acid was added (10.70 mL) drop wise over a period of 30 min at 200C. The reaction mixture was stirred further for 2 hr. The solid formed was filtered, washed with cold acetone (50 mL) and dried. Yield (crude) 59 g, 90.77%.
Example 14: Purification of endo-hexahydro-8-(3-indolylcarbonyIoxy)-2,6- methano-2H-quinolizin-3(4H)-one mesylate, Dolasetron mesylate.
Dolasetron mesylate crude (59 g) was dissolved in hot 5% aqueous isopropanol (500 mL) treated with charcoal and filtered hot. Diethyl ether (50 mL) was added to the filtrate, the solid formed was filtered and dried. Yield 50 g, 82.71%. Purity: 99.9% (HPLC). Example 15: Preparation of endo-hexahydro-8-(3-indolylcarbonyloxy)-2,6- methano-2H-quinoIizin-3(4H)-one mesylate hydrate.
To Dolasetron base (119 g, 0.368 moles) (Step VIII) was dissolved in acetone (2023 mL) and treated with activated charcoal (12 g). Filtered the mixture through hyflow and the clear solution was treated with water (24 ml) and methane sulphonic acid (38.96 g, 0.405 moles) at 25-30°C. The reaction mass was stirred further for 2 h at 0-50C. The solid formed was filtered, washed with acetone (3 x 120 mL) and dried. Yield (crude) 140 g, 87%.
Example 16: Purification of endo-hexahydro-8-(3-indoIylcarbonyloxy)-2,6- methano-2H-quinolizin-3(4H)-one mesylate, Dolasetron mesylate hydrate.
The Dolasetron mesylate (140 g) (Step IX) was taken in water (900 ml) and extracted with ethyl acetate (3x280 ml). The aqueous layer was separated, basified with 10% sodium carbonate (320 mL). The solid obtained was filtered, washed with water and dried. This solid was dissolved in acetone (2 xlOO mL) and treated with activated charcoal (12 g). Filtered the mixture through hyfiow and clear solution was treated with water (20 mL) and methane sulphonic acid (32.72 g, 0.341 moles) at 25-300C. The reaction mass
was stirred further for 2 h at 0-50C. The solid formed was filtered, washed with acetone (3 xlOO niL) and dried. Yield 130 g, 93%. Purity: 99.9% (HPLC).
PREPARATION OF POLYMORPHIC FORMS OF DOLASETRON MESYLATE
Preparation of Dolasetron mesylate Form I
Example 17
0.5g of Dolasetron mesylate was dissolved in 30 mL of IPA at reflux temperature. The hot solution was allowed to cool to room temperature. The solution was stirred at the same temperature for 3 hr. The solid obtained was filtered and dried at 650C to get Dolasetron mesylate Form I. (Kf= 4.85%) Example 18
0.5g of Dolasetron mesylate was dissolved in 20 mL of acetonitrile at reflux temperature. The hot solution was allowed to cool to room temperature. The solution was stirred at the same temperature for 3 hr. The solid obtained was filtered and dried at 65°C to get Dolasetron mesylate Form I. (Kf = 4.56%)
Example 19
0.5g of Dolasetron mesylate was dissolved in 20 mL of acetone at reflux temperature. The hot solution was allowed to cool to room temperature. The solution was stirred at the same temperature for 3 hr. The solid obtained was filtered and dried at 650C to get Dolasetron mesylate Form I. (Kf = 5.32%)
Example 20 0.5g of Dolasetron mesylate was dissolved in 50 mL of ethyl acetate at reflux temperature. The hot solution was allowed to cool to room temperature. The solution was stirred at the same temperature for 3 hr. The solid obtained was filtered and dried at 650C to get Dolasetron mesylate Form I. (Kf= 4.96%)
Example 21
0.5g of Dolasetron mesylate was dissolved in 2 mL of DMF at room temperature. To this clear solution 10 mL of THF was added. The solution was stirred at the same temperature for 3 hr. The solid obtained was filtered and dried at 650C to get Dolasetron mesylate Form I. (Kf = 4.81 %)
Example 22
0.5g of Dolasetron mesylate was dissolved in 2 mL of DMF at room temperature. To this clear solution 10 mL of toluene was added. The solution was stirred at the same temperature for 3 hr. The solid obtained was filtered and dried at 65°C to get Dolasetron mesylate Form I. (Kf = 5.28%)
Example 23
0.5g of Dolasetron mesylate was dissolved in 2 mL of DMF at room temperature. To this clear solution 10 mL of IPA was added. The solution was stirred at the same temperature for 3 hr. The solid obtained was filtered and dried at 65°C to get Dolasetron mesylate Form I. (Kf - 4.75%)
Example 24 0.5g of Dolasetron mesylate was dissolved in 2 mL of DMSO at room temperature.
To this clear solution 10 mL of THF was added. The solution was stirred at the same temperature for 3 hr. The solid obtained was filtered and dried at 650C to get Dolasetron mesylate Form I. (Kf = 5.32%)
Example 25
0.5g of Dolasetron mesylate was dissolved in 2 mL of DMSO at room temperature. To this clear solution 10 mL of toluene was added. The solution was stirred at the same temperature for 3 hr. The solid obtained was filtered and dried at 650C to get Dolasetron mesylate Form I. (Kf = 5.68%)
Example 26
0.5g of Dolasetron mesylate was dissolved in 2 mL of DMSO at room temperature. To this clear solution 10 mL of IPA was added. The solution was stirred at the same temperature for 3 hr. The solid obtained was filtered and dried at 650C to get Dolasetron mesylate Form I. (Kf = 4.52%)
Example 27
0.5g of Dolasetron mesylate was dissolved in 2 mL of N,N-dimethyl acetamide at room temperature. To this clear solution 10 mL of THF was added. The solution was
stirred at the same temperature for 3 hr. The solid obtained was filtered and dried at 650C to get Dolasetron mesylate Form I. (Kf = 4.95%)
Example 28 0.5g of Dolasetron mesylate was dissolved in 2 mL of N,N-dimethyl acetamide at room temperature. To this clear solution 10 mL of toluene was added. The solution was stirred at the same temperature for 3 hr. The solid obtained was filtered and dried at 650C to get Dolasetron mesylate Form I. (Kf = 5.12%)
Example 29
0.5g of Dolasetron mesylate was dissolved in 2 mL of N, N-dimethyl acetamide at room temperature. To this clear solution 10 mL of IPA was added. The solution was stirred at the same temperature for 3 hr. The solid obtained was filtered and dried at 650C to get Dolasetron mesylate Form I. (Kf= 4.82%)
Preparation of Dolasetron mesylate Form II
Example 30
0.5g of Dolasetron mesylate was dissolved in 5 mL of methanol at reflux temperature. The hot solution was allowed to cool to 50C. The solution was stirred at the same temperature for 12 hr. The solid obtained was filtered and dried at 65°C to get Dolasetron mesylate Form II. (Kf = 2.60%)
Preparation of Dolasetron mesylate Form III
Example 31
0.5g of Dolasetron mesylate was dissolved in 10 mL of ethanol at reflux temperature. The hot solution was allowed to cool to 5°C for 2 hr and the solution was stirred at the same temperature for 12 hr. The solid obtained was filtered and dried at 650C to get Dolasetron mesylate Form III. (Kf = 5.48%)
Example 32
0.5g of Dolasetron mesylate was dissolved in 20 mL of ethanol at reflux temperature. The hot solution was allowed to cool to 45°C and to this was added 50 mL of n-hexane. The resultant slurry was stirred for 2 hr and the solution was stirred at the same temperature for 3 hr. The solid obtained was filtered and dried at 650C to get Dolasetron mesylate Form III. (Kf = 4.81%).
Preparation of Dolasetron mesylate Form IV
Example 33
0.5g of Dolasetron mesylate was dissolved in 5 mL of n-propanol at reflux temperature. The hot solution was allowed to cool to 5°C for 2 hr and the solution was stirred at the same temperature for 12 hr. The solid obtained was filtered and dried at 65°C to get Dolasetron mesylate Form IV. (Kf= 5.66%).
Preparation of Dolasetron mesylate Form V
Example 34
0.5g of Dolasetron mesylate was dissolved in 75 mL of chloroform at reflux temperature. The hot solution was maintained at the same temperature for 30 min. The hot solution was allowed to cool to room temperature and was stirred for 3 hr at the same temperature. The solid obtained was filtered and dried at 650C to get Dolasetron mesylate
Form V. (Kf = 5.83%)
Example 35 0.5g of Dolasetron mesylate was dissolved in 75 mL of methylene dichloride at reflux temperature. The hot solution was maintained at the same temperature for 30 min.
The hot solution was allowed to cool to room temperature and was stirred for 3 hr at the same temperature. The solid obtained was filtered and dried at 65°C to get Dolasetron mesylate Form V. (Kf = 7.69%)
Preparation of Dolasetron mesylate Form VI
Example 36
0.5g of Dolasetron mesylate was dissolved in 2 mL of DMF at room temperature. To this solution 20 mL of 1,4-dioxane was added at room temperature. The solution was stirred at the same temperature for 3 hr. The solid obtained was filtered and dried at 65°C to get Dolasetron mesylate Form VI. (Kf = 4.69%) Example 37
0.5g of Dolasetron mesylate was dissolved in 2 mL of DMSO at room temperature. To this solution 10 mL of 1,4-dioxane was added at room temperature. The solution was stirred at the same temperature for 3 hr. The solid obtained was filtered and dried at 650C to get Dolasetron mesylate Form VI. (Kf = 4.46%)
Preparation of Dolasetron mesylate Form VII
Example 38
0.5g of Dolasetron mesylate was dissolved in 2 mL of N,N-dimethyl acetamide at room temperature. To this solution 20 mL of 1,4-dioxane was added at room temperature. The solution was stirred at the same temperature for 3 hr. The solid obtained was filtered and dried at 650C to get Dolasetron mesylate Form VII. (Kf = 4.80%)
Preparation of Dolasetron mesylate Form VIII
Example 39
0.5g of Dolasetron mesylate was suspended in 20 mL of ethyl methyl ketone and the solution was heated to reflux for 1 hr. The solution was cooled to room temperature. The solution was stirred at the same temperature for 3 hr. The solid obtained was filtered and dried at 65°C to get Dolasetron mesylate Form VIII. (Kf = 8.01%)
Preparation of Dolasetron mesylate Form IX
Example 40 0.5g of Dolasetron mesylate was suspended in 20 mL of ethanol and the solution was heated to reflux for 1 hr. The solution was cooled to 45°C temperature. To this hot solution, 50 mL of diisopropyl ether was added and the solution was stirred at 3O0C for 3 hr. The solid obtained was filtered and dried at 65°C to get Dolasetron mesylate Form IX. (Kf = 5.04%). Preparation of amorphous form of Dolasetron mesylate
Example 41
0.5g of Dolasetron mesylate was suspended in 20 mL of methanol, The solution was warmed to 600C to get a clear solution. The hot solution was distilled under vacuum at 55-600C under pressure. The solid obtained by this method was maintained at same temperature and pressure to remove any traces of solvent. The solid obtained was amorphous form of Dolasetron mesylate. (Kf= 4.81%) Example 42
0.5g of Dolasetron mesylate was suspended in 10 mL of water. The solution was warmed to 6O0C to get a clear solution. The hot solution was distilled under vacuum at 55- 600C under pressure. The solid obtained by this method was maintained at same
temperature and pressure to remove any traces of solvent. The solid obtained was amorphous form of Dolasetron mesylate. (Kf = 4.96%) Example 43
0.5g of Dolasetron mesylate was suspended in clean and dry 100 mL round bottom flask and the flask was heated to 16O0C to melt the solid in to liquid. The melt solid was heated to 120° for 3 hr. The solid obtained was amorphous form of Dolasetron mesylate. (Kf = 4.92%) Example 44
A 50 mL aqueous solution of Dolasetron mesylate was frozen using dry ice bath and dried by lyophilization for 24 hr. Example 45
A 5OmL aqueous solution of Dolasetron mesylate at a concentration of 20% weight/volume and at a temperature of 300C was spray dried by a spray gun (PSD 00 Pilot, Hemraj, India at pressure 500 to 600 psi and flow rate of 2 L/hr) at an inlet temperature of 165 C and outlet temperature of 105 C of the spray gun. Example 46
The procedure of Example 45 was carried out at inlet temperature of 155 C and outlet temperature of 950C of the spray gun.
Claims
1. A process for producing polymorphic Form I of Dolasetron mesylate comprising: dissolving Dolasetron mesylate in a solubilizing solvent at a temperature in the range of 300C to 800C, cooling at a temperature in the range of 00C to 300C, isolating and drying the product at a temperature in the range of 300C to 900C.
2. The process as claimed in claim 1 , wherein the solubilizing solvent is selected from isopropanol, acetonitrile, ethyl acetate, acetone or mixture thereof.
3. A process for producing polymorphic Form I of Dolasetron mesylate comprising: dissolving Dolasetron mesylate in a solubilizing solvent, adding an anti-solvent, stirring the suspension with or without cooling for about one hour to about eight hours, isolating and drying the product at a temperature range of 300C to 9O0C.
4. The process as claimed in claim 3, wherein the solubilizing solvent is selected from polar aprotic solvents.
5. The process as claimed in claim 4, wherein the polar aprotic solvent is selected from dimethyl formamide, dimethyl sulfoxide, N, N-dimethyl acetamide or mixture thereof.
6. The process as claimed in claim 3, wherein the anti-solvent is selected from a group of tetrahydrofuran, toluene, isopropanol or mixture thereof.
7. A crystalline polymorphic Form II of endo-hexahydro-8-(3-indolylcarbonyloxy)- 2,6-methano-2H-quinolizin-3(4H)-one methanesulfonate, Dolasetron mesylate characterized by the X-ray powder diffraction pattern as given below:
Peaks in the powder X-ray diffraction pattern are at about 2Θ: 8.5677, 14.3549, 15.3566, 16.1189, 17.4727, 17.9848, 18.2788, 19.9142, 20.3327, 20.7258, 21.4927, 24.4864, 27.5344 and 28.2995 ± 0.2 degrees.
8. A process for producing polymorphic Form II of Dolasetron mesylate of claim 7 comprising: dissolving Dolasetron mesylate in methanol at a temperature in the range of 3O0C to 8O0C, cooling to a temperature in the range of 2°C to 7°C, isolating a residue and drying the residue at a temperature in the range of 300C to 900C.
9. A crystalline polymorphic Form III of endo-hexahydro-8-(3-indolylcarbonyloxy)- 2,6-methano-2H-quinolizin-3(4H)-one methanesulfonate, Dolasetron mesylate characterized by the X-ray powder diffraction pattern as given below:
Peaks in the powder X-ray diffraction pattern are at about 2Θ: 6.9327, 12.6509, 13.7701, 16.6416, 17.5613, 18.1133, 19.4364, 19.6530, 20.5969, 21.2122,
22.1215, 22.3203, 22.9997, 27.9040 and 29.5991 ± 0.2 degrees.
10. A process for producing polymorphic Form III of Dolasetron mesylate of claim 9 comprising: dissolving Dolasetron mesylate in ethanol at a temperature in the range of 300C to 800C, cooling to a temperature in the range of -50C to 300C, isolating a product and diying the product at a temperature in the range of 3O0C to 900C.
11. A process for producing polymorphic Form III of Dolasetron mesylate of claim 9 comprising: dissolving Dolasetron mesylate in a solubilizing solvent at an elevated temperature in the range of 6O0C to 800C, adding an anti-solvent at a temperature in the range of 3O0C to 55°C, isolating a product and drying the product at a temperature in the range of 30°C to 90°C.
12. The process as claimed in claim 11, wherein the solubilizing solvent is selected from methanol, ethanol, n-propanol and isopropanol.
13. The process as claimed in claim 11, wherein the solubilizing solvent is ethanol.
14. The process as claimed in claim 11, wherein the anti-solvent is an aliphatic hydrocarbon.
15. The process as claimed in claim 14, wherein the aliphatic hydrocarbon is selected from n-pentane, n-hexane and n-heptane.
16. The process as claimed in claim 14, wherein the aliphatic hydrocarbon is n-hexane.
17. A crystalline polymorphic Form IV of endo-hexahydro-8-(3-indolylcarbonyloxy)- 2,6-methano-2H-quinolizin-3(4H)-one methanesulfonate, Dolasetron mesylate characterized by the X-ray powder diffraction pattern as given below:
Peaks in the powder X-ray diffraction pattern are at about 2Θ: 8.9231, 12.1275, 12.5348, 12.7100, 14.0712, 14.8069, 16.6676, 17.3884, 18.9666, 19.2953, 19.9217, 20.8115, 22.0471 and 22.4082 ± 0.2 degrees.
18. A process for producing polymorphic Form IV of Dolasetron mesylate of claim 17 comprising: dissolving Dolasetron mesylate in n-propanol at a temperature in the range of 300C to 1000C, cooling to a temperature in the range of -5°C to 3O0C, isolating a product and drying the product at a temperature in the range of 300C to 900C.
19. A crystalline polymorphic Form V of endo-hexahydro-8-(3-indolylcarbonyloxy)- 2,6-methano-2H-quinolizin-3(4H)-one methanesulfonate, Dolasetron mesylate characterized by the X-ray powder diffraction pattern as given below:
Peaks in the powder X-ray diffraction pattern are at about 2Θ: 7.0234, 9.4499, 11.2346, 12.3990, 13.5899, 14.2116, 14.9901, 16.4007, 18.7313, 20.3109, 20.7126, 21.2339, 22.2956, 23.8661, 24.2519, 24.8389, 28.8706 ± 0.2 degrees.
20. A process for producing polymorphic Form V of Dolasetron mesylate of claim 19 comprising: dissolving Dolasetron mesylate in a solubilizing solvent at a temperature at a temperature in the range of 3O0C to 800C, cooling to a temperature in the range of -50C to 3O0C, isolating a product and drying the product at a temperature in the range of 3O0C to '900C.
21. The process as claimed in claim 20, wherein the solubilizing solvent is a chlorinated hydrocarbon.
22. The process as claimed in claim 20, wherein the chlorinated hydrocarbon is either chloroform or methylene dichloride.
23. A crystalline polymorphic Form VI of endo-hexahydro-8-(3-indolylcarbonyloxy)- 2,6-methano-2H-quinolizin-3(4H)-one methanesulfonate, Dolasetron mesylate characterized by the X-ray powder diffraction pattern as given below:
Peaks in the powder X-ray diffraction pattern are at about 2Θ: 7.4433, 9.5049, 9,9496, 10.5039, 12.6288, 13.3801, 13.7613, 16.6316, 17.8499, 19.0352, 19.3432, 20.4909 and 21.8514 + 0.2 degrees.
24. A process for producing polymorphic Form VI of Dolasetron mesylate of claim 23 comprising: dissolving Dolasetron mesylate in a solubilizing solvent at a temperature in the range of 2O0C to 35°C, adding an anti-solvent at a temperature range of 200C to 450C, isolating and drying the product at a temperature in the range of 3O0C to 900C.
25. The process as claimed in claim 24, wherein the solubilizing solvent is a polar aprotic solvent.
26. The process as claimed in claim 25, wherein the polar aprotic solvent is either dimethyl formamide or dimethyl sulfoxide.
27. The process as claimed in claim 24, wherein the anti-solvent is 1, 4-dioxane.
28. A crystalline polymorphic Form VII of endo-hexahydro-8-(3- indolylcarbonyloxy)-2,6-methano-2H-quinolizin-3(4H)-one methanesulfonate, Dolasetron mesylate, characterized by the X-ray powder diffraction pattern as given below:
Peaks in the powder X-ray diffraction pattern are at about 2Θ: 9.4384, 12.5519, 13.3180, 16.5925, 17.2066, 17.7820, 18.9901, 19.2914, 20.5000, 20.9102,
21.2835, 21.7867, 22.0779, 22.7162, 27.1538 and 29.0636 ± 0.2 degrees.
29. A process for producing polymorphic Form VII of Dolasetron mesylate of claim 28 comprising: dissolving Dolasetron mesylate in N,N-dimethyl acetamide at a temperature in the range of 200C to 35°C, adding an anti-solvent at a temperature in the range of 200C to 45°C, isolating and drying the product at a temperature in the range of 300C to 9O0C.
30. The process as claimed in claim 29, wherein the anti-solvent is 1, 4-dioxane.
31. A crystalline polymorphic Form VIII of endo-hexahydro-8-(3- indolylcarbonyloxy)-2,6-methano-2H-quinolizin-3 (4H)-one methanesulfonate, Dolasetron mesylate, characterized by the X-ray powder diffraction pattern as given below:
Peaks in the powder X-ray diffraction pattern are at about 2Θ: 9.3846, 12.3236, 14.1731, 15.0400, 16.2205, 18.3240, 20.7258, 21.0127, 22.0929, 22.5425 and 23.7057 + 0.2 degrees.
32. A process for producing polymorphic Form VIII of Dolasetron mesylate of claim 31 comprising: suspending Dolasetron mesylate in ethyl methyl ketone, heating at an elevated temperature in the range of 4O0C to 800C for one hour, stirring the solution with cooling to a temperature in the range of -50C to 300C, isolating a product and drying the product at a temperature in the range of 3O0C to 9O0C.
33. A crystalline polymorphic Form IX of endo-hexahydro-8-(3-indolylcarbonyloxy)- 2,6-methano-2H-quinolizin-3(4H)-one methanesulfonate, Dolasetron mesylate, characterized by the X-ray powder diffraction pattern as given below:
Peaks in the powder X-ray diffraction pattern are at about 2Θ: 8.9186, 9.5043, 9.9342, 12.2006, 13.0074, 13.5809, 14.2218, 15.1831, 19.0277, 19.7369, 20.3022,
21.2529 and 22.5030 ± 0.2 degrees.
34. A process for producing polymorphic Form IX of Dolasetron mesylate of claim 33 comprising: dissolving Dolasetron mesylate in a solubilizing solvent at a temperature in the range of 600C to 800C, adding an anti-solvent at a temperature in the range of 300C to 55°C, isolating and drying the product at a temperature in the range of 3O0C to 9O0C.
35. The process as claimed in claim 34, wherein the solubilizing solvent is selected from a group consisting of methanol, ethanol and n-propanol.
36. The process as claimed in claim 34, wherein the solubilizing solvent is ethanol.
37. The process as claimed in claim 34, wherein the anti-solvent is aliphatic ether.
38. The process as claimed in claim 37, wherein the aliphatic ether is selected from a group consisting of diethyl ether, diisopropyl ether and methyl tert-butyl ether.
39. The process as claimed in claim 37, wherein the aliphatic ether is diisopropyl ether.
40. An amorphous form of endo-hexahydro-8-(3-indolylcarbonyloxy)-2, 6-methano- 2H-quinolizin-3 (4H)-one methanesulfonate (Dolasetron mesylate).
41. A process for preparation of amorphous form of Dolasetron mesylate of claim 40 comprising: dissolving Dolasetron mesylate in polar protic solvents and vacuum evaporating the solution.
42. The process claimed in claim 41, wherein the polar protic solvent is selected from methanol, ethanol, n-propanol and water.
43. . A process for preparation of amorphous form of Dolasetron mesylate of claim 40 comprising: dissolving Dolasetron mesylate in polar protic solvents and lyophilizing the solution.
44. The process as claimed in claim 43, wherein the polar protic solvent is selected " from methanol, ethanol, n-propanol and water.
45. The process as claimed in claim 43, wherein the polar protic solvent is water.
46. A process for preparation of amorphous form of Dolasetron mesylate of claim 40, comprising: dissolving Dolasetron mesylate in polar protic solvents and spray drying the solution.
47. The process as claimed in claim 46, wherein the polar protic solvent is selected from methanol, ethanol, n-propanol and water.
48. The process as claimed in claim 46, wherein the solution is spray dried at an inlet temperature in the range of 1200C to 1800C.
49. The process as claimed in claim 46, wherein the solution is spray dried at an inlet temperature in the range of 155° C to 165°C.
50. The process as claimed in claim 46, wherein the solution is spray dried at an outlet temperature in the range of 600C to 1100C.
51. The process as claimed in claim 46, wherein the solution is spray dried at an outlet temperature in the range of 950C to 1050C.
52. A process for preparation of amorphous form of Dolasetron mesylate of claim 40 comprising: melting Dolasetron mesylate at a temperature in the range of 15O0C to 1700C and cooling the melt at a temperature in the range of 25°C to 450C.
53. A process for preparation of amorphous form of Dolasetron mesylate of claim 40 comprising: dissolving Dolasetron mesylate in water and lyophilizing the solution.
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IN1610/MUM/2005 | 2005-12-23 | ||
| IN1610MU2005 | 2005-12-23 | ||
| IN1635MU2005 | 2005-12-29 | ||
| IN1635/MUM/2005 | 2005-12-29 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2007072506A2 true WO2007072506A2 (en) | 2007-06-28 |
| WO2007072506A3 WO2007072506A3 (en) | 2008-05-08 |
Family
ID=38189088
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IN2006/000500 WO2007072507A2 (en) | 2005-12-23 | 2006-12-22 | Polymorphic forms of dolasetron base and processes of preparing dolasetron base, its polymorphic forms and salt thereof |
| PCT/IN2006/000499 WO2007072506A2 (en) | 2005-12-23 | 2006-12-22 | Polymorphic forms of dolasetron mesylate and processes thereof |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IN2006/000500 WO2007072507A2 (en) | 2005-12-23 | 2006-12-22 | Polymorphic forms of dolasetron base and processes of preparing dolasetron base, its polymorphic forms and salt thereof |
Country Status (2)
| Country | Link |
|---|---|
| US (1) | US20080275241A1 (en) |
| WO (2) | WO2007072507A2 (en) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7608714B2 (en) | 2006-01-05 | 2009-10-27 | TEVA Gyógyszergyár Zártkörúen Müködö Részvénytársaság | Production of dolasetron |
| CN103360392A (en) * | 2013-06-21 | 2013-10-23 | 辽宁海思科制药有限公司 | Dolasetron mesylate compound |
| CN109503580A (en) * | 2019-01-15 | 2019-03-22 | 南京恩泰医药科技有限公司 | A kind of dolasetron mesilate crystal form and preparation method |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20090030207A1 (en) * | 2007-07-20 | 2009-01-29 | Janos Hajko | Polymorphs of Dolasetron base and process for preparation thereof |
| CN102183589B (en) * | 2010-07-02 | 2013-04-24 | 成都新恒创药业有限公司 | Method for detecting contents of dolasetron isomer and salt thereof |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0329905A1 (en) * | 1988-02-23 | 1989-08-30 | Merrell Dow Pharmaceuticals Inc. | Use of quinolizine and quinolizinone derivatives in the manufacture of medicaments for increasing gastric motility |
| EP0330824A1 (en) * | 1988-03-01 | 1989-09-06 | Merrell Dow Pharmaceuticals Inc. | Use of quinolizine and quinolizinone derivatives in the manufacture of medicaments for the treatment of cardiac arrhythmias |
| EP0339669A1 (en) * | 1988-04-29 | 1989-11-02 | Merrell Dow Pharmaceuticals Inc. | Process for preparing indole-3-carboxylic acid esters of trans-hexahydro-8-hydroxy-2,6-methano-2H-quinolizin-3(4H)-one |
| US4906755A (en) * | 1986-11-03 | 1990-03-06 | Merrell Dow Pharmaceuticals Inc. | Esters of hexahydro-8-hydroxy-2,6-methano-2H-quinolizin-3-(4H)-one and related compounds |
| WO2006026927A1 (en) * | 2004-09-10 | 2006-03-16 | Chengdu Giantech Hi-Technology Development Co., Ltd. | A new polymorph of dolasetron mesylate monohydrate and preparation thereof |
| WO2006056081A1 (en) * | 2004-11-25 | 2006-06-01 | Cilag Ltd. | Method for preparing hexahydro-8-hydroxy-2,6-methano-2h-chinolizin-3(4h)-one esters |
-
2006
- 2006-12-22 WO PCT/IN2006/000500 patent/WO2007072507A2/en active Application Filing
- 2006-12-22 WO PCT/IN2006/000499 patent/WO2007072506A2/en active Application Filing
- 2006-12-22 US US12/158,708 patent/US20080275241A1/en not_active Abandoned
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4906755A (en) * | 1986-11-03 | 1990-03-06 | Merrell Dow Pharmaceuticals Inc. | Esters of hexahydro-8-hydroxy-2,6-methano-2H-quinolizin-3-(4H)-one and related compounds |
| EP0329905A1 (en) * | 1988-02-23 | 1989-08-30 | Merrell Dow Pharmaceuticals Inc. | Use of quinolizine and quinolizinone derivatives in the manufacture of medicaments for increasing gastric motility |
| EP0330824A1 (en) * | 1988-03-01 | 1989-09-06 | Merrell Dow Pharmaceuticals Inc. | Use of quinolizine and quinolizinone derivatives in the manufacture of medicaments for the treatment of cardiac arrhythmias |
| EP0339669A1 (en) * | 1988-04-29 | 1989-11-02 | Merrell Dow Pharmaceuticals Inc. | Process for preparing indole-3-carboxylic acid esters of trans-hexahydro-8-hydroxy-2,6-methano-2H-quinolizin-3(4H)-one |
| WO2006026927A1 (en) * | 2004-09-10 | 2006-03-16 | Chengdu Giantech Hi-Technology Development Co., Ltd. | A new polymorph of dolasetron mesylate monohydrate and preparation thereof |
| WO2006056081A1 (en) * | 2004-11-25 | 2006-06-01 | Cilag Ltd. | Method for preparing hexahydro-8-hydroxy-2,6-methano-2h-chinolizin-3(4h)-one esters |
Non-Patent Citations (1)
| Title |
|---|
| GU C-H ET AL: "Grouping solvents by statistical analysis of solvent property parameters: implication to polymorph screening" INTERNATIONAL JOURNAL OF PHARMACEUTICS, AMSTERDAM, NL, vol. 283, no. 1-2, 28 September 2004 (2004-09-28), pages 117-125, XP004560638 ISSN: 0378-5173 * |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7608714B2 (en) | 2006-01-05 | 2009-10-27 | TEVA Gyógyszergyár Zártkörúen Müködö Részvénytársaság | Production of dolasetron |
| CN103360392A (en) * | 2013-06-21 | 2013-10-23 | 辽宁海思科制药有限公司 | Dolasetron mesylate compound |
| CN109503580A (en) * | 2019-01-15 | 2019-03-22 | 南京恩泰医药科技有限公司 | A kind of dolasetron mesilate crystal form and preparation method |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2007072507A3 (en) | 2008-01-24 |
| WO2007072507A2 (en) | 2007-06-28 |
| US20080275241A1 (en) | 2008-11-06 |
| WO2007072506A3 (en) | 2008-05-08 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR101440257B1 (en) | Process for the manufacture of intermediates for preparing pharmaceutically active compounds | |
| AU2006281449A1 (en) | Method for producing betamimetics | |
| CA2698245A1 (en) | Process and intermediates for preparing integrase inhibitors | |
| US20060148866A1 (en) | Novel process for preparing pramipexole and its optical isomeric mixture by reduction with sodium triacetoxyborohydride | |
| JP6081450B2 (en) | Crystalline salt of asenapine | |
| US20070203176A1 (en) | Crystalline forms of dolasetron base and processes for preparation thereof | |
| JP6901976B2 (en) | How to prepare xanthine-based compounds | |
| WO2007072506A2 (en) | Polymorphic forms of dolasetron mesylate and processes thereof | |
| CN110891947B (en) | Process for preparing ailutinib or a pharmaceutically acceptable salt thereof | |
| US20080200681A1 (en) | Processes for preparing palonosetron salts | |
| WO2010015107A1 (en) | Stereoselective synthesis of valiolamine | |
| WO2023143630A1 (en) | Preparation method for nucleoside analogue vv116 | |
| WO2014111954A1 (en) | Process for the preparation and purification of apixaban | |
| CA2960473A1 (en) | Processes for the preparation of tadalafil and intermediates thereof | |
| CN110177790B (en) | Resolution of optically active diazaspiro [4.5] decane derivatives | |
| EP1551414B1 (en) | Process for preparing the crystal form I of olanzapine | |
| Brine et al. | An improved resolution of (±)‐cis‐N‐normetazocine | |
| EP3911660B1 (en) | Process for preparation of 2-amino-5-hydroxy propiophenone | |
| US8044196B2 (en) | Process for producing pure form of 2-methyl-4-(4-methyl-1-piperazinyl)-10H-thieno[2,3-b][1,5]benzodiazepine | |
| CN108586450B (en) | Recrystallization purification method of choline M receptor anticaking agent | |
| CN113214164B (en) | Synthetic method of intermediate 2,4-dimethylpyrimidin-5-ol of Lebo Lei Sheng | |
| JP2009526030A (en) | CABERGOLINE AND METHOD FOR PRODUCING NOVEL POLYMORPHIM FORM | |
| CN115703769A (en) | Preparation method of azilsartan | |
| CN112110910A (en) | Method for preparing rivaroxaban intermediate and method for preparing rivaroxaban by using same | |
| JPH0234346B2 (en) |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 06848719 Country of ref document: EP Kind code of ref document: A2 |